











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La fibroelastosis pleuroparenquimatosa (FEPP) pertenece al grupo de enfermedades pulmonares intersticiales difusas (EPID) raras, que se caracteriza por fibrosis de la pleura visceral y del parénquima pulmonar subyacente de los lóbulos superiores1. No se conoce su incidencia ni prevalencia aunque en una serie de 1622 pacientes con EPID y 205 biopsias, se relata que el 5,9% correspondía a casos de FEPP2.
Inicialmente Davies y cols relataron cinco casos de enfermedad pulmonar fibrosante progresiva en los cuales la afectación estaba confinada a las regiones superiores y que los autores asimilaron a aquellas lesiones vistas en la espondilitis anquilosante3. Más adelante, Amitani y cols publica una serie de 13 pacientes de características similares, acuñando el nombre de fibrosis pulmonar de lóbulo superior o PULF (siglas en inglés)4, aunque en el 2004 Frenkel y cols. propusieron el nombre de fibroelastosis pleuroparenquimatosa o FEPP5. Recién en el 2013 se encuentra en la clasificación multidisciplinaria internacional de las neumonías intersticiales idiopáticas6. En el Brasil y en la Argentina ya se describieron los primeros casos7,8.
Existen escasos reportes sobre la prevalencia de hipertensión pulmonar en la FEPP y existen pocos reportes al respecto. Hasta la fecha no fueron publicados casos de diagnóstico de esta enfermedad en nuestro país.
PRESENTACION DE CASO
Paciente de sexo masculino de 26 años, procedente de área urbana de Luque, no fumador y sin comorbilidades que presenta cuatro años de historia de falta de aire lentamente progresiva acompañado de tos seca e intermitente. No consta exposición a químicos ni tóxicos. Recurre a varios centros públicos y privados, siendo internado hace dos años en uno de ellos y manejado como tuberculosis pulmonar, sin respuesta al tratamiento anti bacilar. Existen referencias de una segunda internación ocho meses atrás, en cuyo resumen de egreso se menciona hipótesis diagnostica de neumonía de lenta resolución, y durante la que se realiza lavado broncoalveolar de cuyo cultivo se obtiene Aspergillus sp por lo que se procede a tratamiento con voriconazol, también sin respuesta clínica.
Un mes antes de la consulta al Hospital General de Luque presenta empeoramiento de la disnea hasta hacerse en reposo sumado a expectoración amarillenta en moderada cantidad. No presenta cuadro respiratorio exacerbado durante los años de pandemia y tiene dos dosis de vacunas específicas para COVID-19. Es traído al Servicio de Urgencias por historia de 24 hs de aumento en la dificultad respiratoria y dolor tipo puntada en hemitorax izquierdo no acompañado de fiebre ni chillido de pecho.
Ingresa pálido y sudoroso con los siguientes signos vitales: presión arterial 114/80 mmHg; frecuencia cardiaca 117 x min; frecuencia respiratoria 45 x min; temperatura axilar 37,5°C, saturación O2: 90%en vigencia de oxigenoterapia por cánula nasal a 5L/min.
El examen físico del aparato respiratorio denota disminución del diámetro anteroposterior del tórax (platitórax) con profundización del hueco supraesternal y taquipnea, tiraje universal configurando una mala mecánica respiratoria, notándose disminución de murmullo vesicular en campos pulmonares superiores y medios con crepitantes y escasas sibilancias en ambas bases.
Exámenes auxiliares: hemoglobina 10g/dl, glóbulos blancos 17.850/mm3 con 79% de neutrófilos, glicemia pos-prandial: 124mg/dl, dímero D 0,25pg/ml (<0,5), ferritina 310ng/ml (<300), procalcitonina 0,1ng/ml (<0,5), lactato 25,2mg/dl (4,5-19,8), pro-BNP 800 pg/ml. Orina simple sin datos llamativos. Gasometría arterial (oxigenoterapia en vigencia): pH 7,41, pCO2:40,4 mmHg, pO2: 104mmHg, HCO3: 25mEq/L, SatO2: 91%. Rx de tórax: opacidades corticales bilaterales en región superior y media. Electrocardiograma: desvío del eje a derecha y extrasístoles supraventriculares aisladas. Hisopado nasofaríngeo (reacción en cadena de polimerasa): negativo para Influenza A, influenza H1N1, influenza estacional, influenza H3N2, influenza B, virus sincitial respiratorio, rinovirus, coronavirus SARS-CoV-2, parainfluenza, metapneumovirus, adenovirus, bocavirus, enterovirus, Chlamydophilla pneumonia, Legionella pneumophila, Mycoplasma pneumoniae, Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarralhis, Bordetella pertussis, Bordetella parapertussis, ANA, factor reumatoide y anti SCL-70 negativos. Ecocardiografia: dilatación de cavidades derechas y presión sistólica de arteria pulmonar estimada de 65mmHg.Tomografía computarizada de tórax: patrón de enfermedad pulmonar intersticial difusa (Figura 1 a-c).
Se solicita interconsulta con Neumología que elabora el diagnóstico de Fibroelastosis pleuroparenquimatosa, hipertensión pulmonar y cor pulmonale. Es trasladado a la Unidad de Cuidados Intensivos por el diagnóstico de insuficiencia respiratoria crónica agudizada donde se procede a intubación orotraqueal y asistencia respiratoria mecánica. Se inicia antibioticoterapia con beta-lactámicos y quinolonas intravenosas posterior a toma de cultivos de varios materiales que resultan sin rédito bacteriológico. Disminución de leucocitosis y mejoría de relación PaO2/FiO2, en los siguientes días, aunque la distensibilidad pulmonar se muestra disminuida persistentemente. La agudización del cuadro y el patrón de perfusión en mosaico más los hallazgos en la ecocardiografía indujeron a pensar en un evento embólico pulmonar que no se evidenció mediante angiotomografía torácica (Figura 1 d-f). Después de varias semanas de empeoramiento progresivo, paciente fallece.
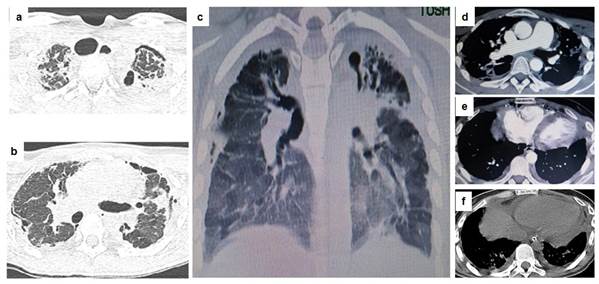
Figura 1. (a) TAC de tórax, corte axial, que denota hueco supra esternal profundizado, opacidades pleurales, neumotórax laminar anterior izquierdo y espesamiento de septos interlobulares bilaterales junto a bronquiectasia de tracción. Destaca traqueomegalia secundaria; (b) TAC de tórax, corte coronal, que muestra el aplanamiento de torax (platitorax) y espesamiento pleural y bronquiectasia de tracción con área de consolidación predominante en lóbulo superior. Se nota vidrio esmerilado y patrón en perfusión en mosaico en áreas dependientes. Se destaca pequeña área de neumotórax contenido en región medial derecha. (c) corte sagital del tórax donde se aprecia vidrio esmerilado, engrosamiento pleural en ambos vértices y aumento de ramos descendentes de la arteria pulmonar; (d) TAC contrastada que denota aumento del diámetro de la arteria pulmonar; (e) ventrículo derecho notoriamente aumentado de tamaño; (f) derrame pericárdico.
DISCUSIÓN
Existen formas idiopáticas y secundarias de FEEP. Se describen casos de asociación con exposición ocupacional a asbesto y aluminio, trasplante de medula ósea o de pulmón, antecedente de quimioterapia (ciclofosfamida y metotrexate) y radiación9 Puede aparecer en compañía de neumonitis intersticiales no clasificables, neumonitis intersticial no específica y neumonitis por hipersensibilidad10. En ocasiones hay antecedente de historia familiar de la enfermedad11 y siempre hay que considerar la posibilidad de concomitancia con enfermedades autoinmunes como esclerodermia, artritis reumatoide, síndrome de Sjoëgren, miopatía inflamatoria idiopático, síndrome de superposición, enfermedad indiferenciada del tejido conjuntivo, granulomatosis con poliangitis y poliangitis microscópica12,13. Gudmunsson y colaboradores encontraron que entre 25 y 36% de pacientes con FPI muestran lesiones que recuerdan a la FEEP14.
Dos epifenómenos se destacan en el caso reportado aquí. En primer lugar, la historia del hallazgo de Aspergillus en el lavado broncoalveolar que puede emerger como complicación según Kurosaki y cols15, aunque existe un relato de lesiones radiológicas seudo-fibroelastosis secundarias a la micosis16. En segundo lugar, debe mencionarse al neumotórax que, si bien no ocupa un relato sintomático aquí, se manifestó como hallazgo radiológico incidental. En una serie de 89 pacientes con FEPP idiopática se constató que el 59,6% había desarrollado agudización por neumotórax y que en la mayoría de estos se constató fuga persistente (fistula) pese al drenaje torácico evacuador proyectando implicancias en la sobrevida. Es relevante mencionar que la frecuencia de neumotórax es mayor en la FEEP que en la FPI17.
En la práctica se sugiere un enfoque multidisciplinar para el diagnóstico teniendo en cuenta criterios clínico-radiológicos y cuando disponibles, datos histopatológicos los cuales frecuentemente son difíciles de obtener en estadios clínicos avanzados. Los diagnósticos diferenciales se establecen con la neumonitis por hipersensibilidad, sarcoidosis, neumointis intersticiales idiopáticas, infección por mycobacterias, neumoconiosis, neoplasias y casquete pleural secuelar18.
La sobrevida promedio de 85 pacientes fue de 11 años en el trabajo de Watanabe y cols. pero recientemente se describieron fenotipos de FEEP progresiva con historia de 2-6 años hasta el fallecimiento19,20. Otro reciente estudio en 36 pacientes encontró que el 33% progresaba desde el diagnóstico hasta el óbito en 12 meses, resultando en una sobrevida promedio de 24 meses21. La FEPP idiopática, el acortamiento de telómeros, patrón de neumonitis intersticial usual concomitante y la neumonitis por hipersensibilidad asociada, confieren factores pronósticos de evolución catastrófica18.
El desarrollo de hipertensión pulmonar agrega un pronóstico adverso a varias EPID entre las que la FPI, la neumonitis por hipersensibilidad fibrosante, las asociadas a enfermedades del colágeno, la sarcoidosis y la histiocitosis de células de Langerhans se citan comúnmente. Es de notar que, debido a su fisiopatología multifactorial, la sarcoidosis y la histiocitosis de células de Langherhans se clasifican en grupos diferentes ya que no se consideraa la hipoxemia como única variable explicadora, sino que otros factores como enfermedades vasculares, o linfadenopatias que distorsionan el mediastino o disfunción ventricular pueden estar inmiscuidas como causales22. La hipertensión pulmonar en las EPID puede acometer en ausencia de hipoxemia en reposo y/o enfermedad avanzada pudiendo notarse una ausencia de correlación entre los niveles de hipertensión pulmonar y el grado de alteraciones en las pruebas de función pulmonar. Varios otros mecanismos pueden estar implicados: disfunción endotelial, estrés oxidativo, vías de paso inmune alterado, fibrosis perivascular y hasta predisposición genética23.
Existen escasos reportes de FEPP asociados a hipertensión pulmonar. Boerner y cols. reportan el caso de un paciente de 68 años con hipertensión pulmonar leve, que pese a tratamiento con pirfenidona, fallece a los cuatro años24. Otros autores relatan el caso de un paciente de 71 años con cateterismo cardiaco derecho con 30 mmHg en presión media de arteria pulmonar25. En 19,3% de una serie de 83 pacientes se constató hipertensión pulmonar y la sobrevida en este grupo fue menor (16,3 meses vs 50,2 meses) que aquellos que no cursaban con alteración hemodinámica26. Khiroya y cols evaluaron 43 biopsias y encontraron espesamiento fibrointimal venoso y arterial de grado variable en más del 90% de las láminas27.
En la hipertensión pulmonar asociada a FPI, la remodelación de los ramos de la arteria pulmonar comprende particularmente al espesamiento de la capa de células musculares y la presencia de lesiones proliferativas de la íntima28. En otra serie retrospectiva de pacientes se encontró una prevalencia de 25% de hipertensión pulmonar en la FEEP. Revisando las láminas de biopsia y comparando a la estructura de las arterias de FPI y pulmones normales, se encontró que la proliferación elástica de la media y el espesamiento de la adventicia con fibras de colágeno fueron características histopatológicas diferenciales de la FEPP23. Este trabajo sugiere un tipo diferenciado de complicación cardiovascular en este grupo de pacientes.
¿Porque algunas EPID desarrollan hipertensión pulmonar y otras no? ¿Existen fenotipos que conllevan mayor riesgo de tener formas más agresivas de hipertensión pulmonar?29 ¿Porque no todas las EPID con hipertensión pulmonar desarrollan Cor pulmonale? Aunque en el caso presentado en este reporte no se comprobó evento de embolia pulmonar, no se puede descartar la concomitancia de hipertensión pulmonar tromboembólica crónica (HPTC). La asociación entre EPID y HPTC se cita aisladamente en sarcoidosis30 y es al parecer frecuente encontrar fenómenos de tromboembolismo venoso en la neumonitis por hipersensibilidad crónica, así como en la fibrosis pulmonar idiopática31.
Aparte de las medidas de soporte y el trasplante pulmonar, no existe tratamiento específico para esta enfermedad. El análisis retrospectivo del uso de antifibróticos en 64 pacientes con FEPP y patrón de neumonitis intersticial usual comparado a 195 sujetos con FPI clásica, demostró que la sobrevida del primer grupo era significativamente menor lo cual propone una limitada una limitada eficacia de los fármacos32. La asociación con hipertensión pulmonar tampoco tiene un tratamiento específico y se necesitan estudios que evalúen procedimientos, fármacos o una combinación de factores que puedan pintar de esperanza el horizonte de estos pacientes. En el Paraguay, queda aún pendiente el inicio de la era de los trasplantes pulmonares.

