










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista del Nacional (Itauguá)
Print version ISSN 2072-8174
Rev. Nac. (Itauguá) vol.6 no.2 Itauguá 2014
CASOS CLÍNICOS
Neumopatia intersticial en pacientes con polimiositis y dermatomiositis: reporte de cuatro casos
Interstitial Lung Disease in Patients With Polymyositis and Dermatomyositis: four Cases Report
Yngrid González2 , Estela Torres1, Dora Montiel1, Laura Bordenave1, Gabriela Cardozo2
RESUMEN
Se reporta cuatro casos: tres con dermatomiositis y uno con poliomiositis con compromiso pulmonar, tratados con pulsos de metilprednisolona asociado con azatioprina en tres casos y uno con ciclofosfamida, tres de ellos fallecieron, dos por complicaciones infecciosas pulmonares, el tercero por insuficiencia respiratoria aguda, Uno solo presento buena evolución bajo tratamiento con inmunoglobulina .En los cuatro pacientes el anticuerpo anti Jo fueron negativos.
Palabras clave: Dermatomiositis, Polimiositis, Neumopatía Intersticial.
ABSTRACT
We report four cases, three with dermatomyositis and polymyositis one with pulmonary involvement treated with pulse methylprednisolone associated with azathioprine in three cases and one cyclophosphamide, three of them died, two infectious pulmonary complications, the third for acute respiratory failure, one presented good performance under treatment with immunoglobulin .In the four patients were negative anti Jo
Keywords: Dermatomyositis, Polymiositis, Interstitial Pneumopathy.
INTRODUCCIÓN:
Las enfermedades del tejido conectivo representan un grupo heterogéneo de trastornos inflamatorios que están mediados inmunológicamente y que pueden afectar a múltiples órganos, entre ellos el pulmón. Dentro de estas enfermedades, la polimiositis y la dermatomiositis pueden comprometer el pulmón en diferentes formas: neumonía aspirativa o insuficiencia ventilatoria como complicación de la debilidad muscular, enfermedad pulmonar secundaria a fármacos o a carcinoma de pulmón y enfermedad pulmonar intersticial1-2-3-4.
La dermatomiositis y la polimiositis conforman el principal grupo de miopatías adquiridas en el adulto, las cuales tienen una incidencia aproximada de 0,6 a por 100.000 personas. Estas enfermedades se caracterizan por debilidad muscular simétrica y proximal de instauración insidiosa, usualmente no dolorosa, con elevación de las enzimas musculares hasta 50 veces por encima de su valor normal, alteraciones evidentes en la biopsia de músculo en la microscopía electrónica y de luz, y por cambios electromiográficos característicos5.
La Polimiositis, es una enfermedad rara que afecta a los adultos. La dermatomiositis afecta a niños y adultos y a mujeres con mayor frecuencia que a varones5.
No es fácil precisar la fecha real de inicio de la polimiositis y de manera típica los pacientes retrasan varios meses la consulta con el médico, esto es diferente de lo que ocurre en la dermatomiositis en que el eritema facilita la identificación temprana. La polimiositis se parece a otras miopatías y es de diagnóstico de exclusión y como entidad aislada es rara; con más frecuencia aparece algún trastorno autoinmunitario generalizado o alguna conjuntivopatía o infección vírica o bacteriana identificada5.
La neumopatía intersticial hoy en día constituye la principal causa de muerte en estos pacientes, a veces antecede a la miopatía o se presenta en la fase temprana de esta enfermedad y ocurre incluso entre el 10 y 40% de los pacientes con polimiositis o dermatomiositis6-7.
Generalmente los pacientes con neumonitis intersticial presentan artralgias, síntomas generales y fiebre hasta en el 80% de los casos; el compromiso miopático está presente en más del 90% y el fenómeno de Raynaud entre el 30% y el 50% de ellos8.
De los individuos con polimiositis y dermatomiositis, el 80% presenta anticuerpos anti Jo positivos. El Anti-Jo1 fue el primero de los anticuerpos antisintetasa caracterizado; se describió en 1976 en el suero del paciente John P (de ahí su nombre), quien debutó con un cuadro de neumonía intersticial asociada a una miopatía inflamatoria. Es el prototipo de anticuerpos cuyo blanco es la histidil-tRNA sintetasa, una enzima citoplasmática que acetila el RNA de trasferencia y es el único utilizado en la práctica clínica9-10.
Varios mecanismos explican la asociación entre la producción de dichos anticuerpos y las miopatías inflamatorias. Se propone que fenómenos de mimetismo molecular con enzimas virales permiten la pérdida de inmunotolerancia. Un ejemplo de esto es el trabajo realizado por Bowles en el cual se buscó virus Coxsackie en pacientes con miopatía inflamatoria, otras enfermedades musculares y controles sanos, encontrando este sólo en el tejido muscular de los pacientes afectados por algunos de los subtipos de miopatía inflamatoria. La unión del RNA viral a la histidil RNA trasferasa humana propiciaría el fenómeno de pérdida de inmunotolerancia. Una vez que se producen anticuerpos anti-Jo1, éstos inducen respuestas celulares y humorales que generan inflamación de las fibras musculares; los complejos inmunes circulantes podrían estimular macrófagos alveolares y favorecer la inflamación. Por el contrario, la expresión enriquecida de Jo-1 en monocapas de células alveolares pulmonares clivadas con granzima B, en comparación con otros tejidos, incluido el músculo, sugiere que el tejido que inicia la respuesta autoinmune en el "síndrome anti-Jo1" es el pulmón, con ataque secundario del músculo11.
Para realizar el diagnóstico también es necesario realizar una Radiografía Simple de tórax, que a veces no evidencia la afección intersticial, pero sí una Tomografía Axial Computada de Alta Resolución de tórax en donde se pueden observar los parches de neumonitis. En la espirometría se observa un patrón restrictivo de la función pulmonar, generalmente precedido por una disminución de la capacidad de difusión de monóxido de carbono (DLCO), que es el método más sensible para detectar el compromiso intersticial inicial y tiene valor pronóstico, pues la presencia de una DLCO disminuida por debajo del 45% en la ausencia de hipertensión pulmonar es un fuerte predictor de cronicidad. Lo que sella el diagnóstico es la biopsia pulmonar ya sea por fibrobroncoscopía o a cielo abierto, pero no se indica en casos en donde la enfermedad pulmonar es importante y evidente por los estudios de imágenes, ya que se debe tener en cuenta el riesgo – beneficio de este procedimiento para el paciente12-13-14.
En cuanto al tratamiento, los esteroides tienen efectos benéficos en las manifestaciones sistémicas y en algunos subtipos de compromiso pulmonar, lo antes posible se debe administrar prednisona 1mg/kg/día y luego de 10 semanas reducir lentamente hasta 1mg/kg en días alternos15. En términos generales, la respuesta pulmonar es tardía con respecto a la mejoría muscular y se obtiene a expensas de altas dosis con sus subsecuentes efectos secundarios, motivo por el cual se ha descrito tratamiento adicional con: pulsos de ciclofosfamida 1g/m2, azatioprina hasta 3mg/kg/día y metotrexate hasta 25mg. por semana. La ciclofosfamida se ha utilizado como terapia de inducción logrando remisión y, en ocasiones, regresión completa de los infiltrados pulmonares. La azatioprina y el metotrexate han mostrado efectividad como ahorradores de esteroides y para detener la progresión de la enfermedad a pesar de presentar recaídas con el desmonte de tratamiento. El metrotexate, por su posible toxicidad pulmonar, es generalmente evitado. La inmunoglobulina intravenosa ha sido estudiada para el manejo de miopatía inflamatoria sin tener un papel claro en el tratamiento de la enfermedad pulmonar intersticial. Späth y colaboradores describieron su experiencia con 12 pacientes con neumonitis intersticial asociado a miopatías, encontrando en dos de ellos una respuesta adecuada; no obstante, debe recordarse el carácter transitorio del efecto de las inmunoglobulinas, pudiendo tener algún papel en el control inicial del cuadro. Por lo general se necesita repetir la administración en goteo intravenoso cada 6 a 8 semanas para conservar la mejoría. Se ha recomendado una dosis de 2g/Kg de peso fraccionada en el transcurso de dos a cinco días por ciclo terapéutico. Al parecer la plasmaféresis y la leucoféresis no son eficaces en el tratamiento de estos pacientes16.
Los que tienen peor pronóstico son los pacientes con afección grave al inicio o tratados con mucho retraso16.
Objetivo: Presentar tres pacientes con dermatomiositis y uno con poliomiositis con compromiso pulmonar.
Caso 1: Varón de 53 años con cuadro de 9 meses, que inició con lesiones eritemato - violáceas, en rostro, tórax, miembros superiores, fotosensibilidad, caída de cabello, artralgias, fiebre, pérdida de peso. 3 meses antes se agrega al cuadro disnea progresiva hasta llegar al reposo. Al examen físico se constata: alopecia difusa, eritema heliotropo, lesiones eritemato violáceas en región malar, tórax, miembros superiores (figura 1), signo de Gottron; con escasa mejoría con Hidroxicloroquina 400mg/día y prednisona 1mg/kp/día. Laboratorialmente se evidencia: Creatinfosfoquinasa 925Ul/l; Aldolasa: 6 UI/l; Anticuerpo Anti Jo, Anticuerpos antinucleares, anti DNA y ELISA para HIV negativos. Gasometría arterial: Alcalosis respiratoria con hipoxemia. Radiografía simple de tórax: infiltrado intersticial difuso bilateral. Tomografía Axial Computada de Alta Resolución de Tórax Simple: Infiltrado intersticial en vidrio esmerilado (figura 2 y 3). Espirometría: incapacidad ventilatoria restrictiva. Ecocardiografía: Presión Sistólica Pulmonar: 45mmHg. Evoluciona a insuficiencia respiratoria aguda, ingresa a la unidad de cuidados intensivos, se realiza 3 pulsos 1g/día por tres días de metilprednisolona asociado a ciclofostamida en pulsos 1g/m2 de superficie corporal, sin mejoría. Paciente fallece.
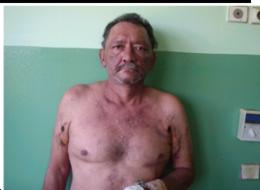
Fig.1. Eritema malar. Signo del Chal.
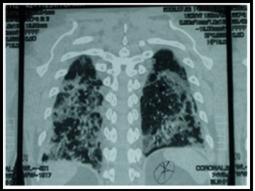
Fig. 2. TACAR simple de tórax. Parches de Neumonitis
Caso 2: Mujer de 43 años, consulta por un cuadro de 1 mes de evolución que inició con lesiones eritemato - violáceas en rostro, tórax, miembros superiores, caída de cabello, pérdida de 4 kg. Concomitantemente presenta tos, disnea progresiva hasta llegar al reposo. Al examen Físico: alopecia difusa, eritema en heliotropo, lesiones eritemato- violáceas en región malar, tórax y miembros superiores; disminución de fuerza muscular proximal 4/5, fenómeno de Raynaud. Laboratorio: Creatinfosfoquinasa: 28 UI/l. ANA, anti DNA, Anti Jo y HIV negativos. Gasometría arterial: alcalosis respiratoria con hipoxemia; Radiografía simple de tórax: infiltrado intersticial en panal de abejas bilateral; Ecocardiografía: Presión Sistólica Pulmonar: 36 mmHg. Tomografía Axial Computada de Alta Resolución de Tórax Simple: Aspecto en vidrio esmerilado difuso, bullas y nódulos pequeños, Espirometría: Incapacidad ventilatoria restrictiva. Se trató a la paciente con pulsos de metil prednisolona 1g/día por tres días seguido de prednisona oral 1mg/kp/día y asociado a azatioprina 2mg/kp/día, presentó evolución a insuficiencia respiratoria aguda, se complica con neumonía intrahospitalaria y fallece en la Unidad de Cuidados Intensivos.
Caso 3: Varón de 27 años, con cuadro de 3 meses, que inició con debilidad muscular proximal, artralgias, fiebre, disnea progresiva hasta llegar al reposo. Examen físico: debilidad muscular proximal de los cuatro miembros 2/5. Laboratorio: Creatinfosfoquinasa: 2556 UI/l, Aldolasa: 45U/l, ANA 1: 1200 patrón moteado fino, Anti DNA: Negativo, Anti Jo y HIV negativos. Gasometria arterial: Normal. Radiografía de tórax: infiltrado intersticial bilateral, Tomografía Axial Computada de Alta Resolución Simple de Tórax: infiltrado vidrio esmerilado difuso. Ecocardiografía: Presión Sistólica Pulmonar: 42mmHg. Espirometría: Incapacidad ventilatoria restrictiva. Tratamiento: pulsos de metil prednisolona 1g/d por tres días, azatioprina 2mg/k/p/día; presentó mejoría total del compromiso muscular y parcial del pulmonar, la disnea se intensifica y se realiza tratamiento con inmunoglobulina humana 400mg/k/p/día durante 5 días, posteriormente pulsos de ciclofosfamida 1g/m2 mensuales, con recuperación completa de la afectación pulmonar con espirometria de control normal a los 9 meses de tratamiento.
Caso 4: Mujer de 59 años, que consulta por cuadro de 20 días de evolución por lesiones eritemato-violáceas a nivel del cuello, escote, miembros superiores y de las manos, mialgias, artralgias, sensación febril y aparición de aftas dolorosas a nivel de mucosa bucal y lengua, a su ingreso se constata disnea en reposo, que se intensifica con el correr de los días. Examen físico: A nivel de piel: Eritema en heliotropo (figura 4), signo de Gottron (figura 5), manos de mecánico (figura 5), signo del Chal. Debilidad muscular proximal de los cuatro miembros 2/5. Lesión ulcerosa en lengua y mucosas. Laboratorio: Creatinfosfoquinasa: 3796 UI/l, Aldolasa: 36, ANA: Negativo. Anti DNA: Negativo. Anti Jo y HIV: negativos. Gasometría arterial: alcalosis respiratoria con hipoxemia. Radiografía de tórax: Infiltrado intersticial bilateral. Tomografía Computada de Alta Resolución de Tórax Simple: Infiltrado intersticial en panal de abejas (figura 6). Ecocardiografía: Presión Sistólica Pulmonar: 38mmHg. Recibe 3 pulsos de metilprednisolona 1g/día y 1 bolo de ciclofosfamida 1g/m2 de superficie corporal, pero el cuadro no mejora y la paciente se complica con una neumonía intrahospitalaria, evoluciona a insuficiencia respiratoria aguda y fallece en la unidad de cuidados intensivos.
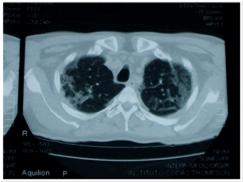
Fig. 3. Infiltrado Intersticial en Vidrio Esmerilado
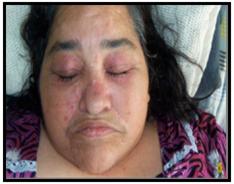
Fig. 4. Eritema en heliotropo
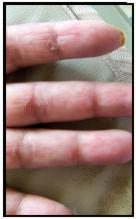
Fig. 5. Manos de mecánico y signo de Gottron
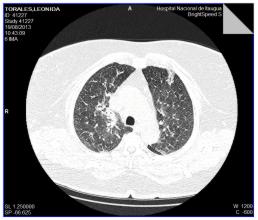
Fig. 6. Infiltrado Intersticial en Panal de Abejas
COMENTARIO
La Neumopatía intersticial se produce en aproximadamente entre el 10 y 40% de los pacientes con dermatomiositis o polimiositis, puede ser de instalación crónica y lentamente progresiva o aguda y rápidamente progresiva1-2.
El diagnóstico por lo general puede ser establecido sobre la base de la presentación clínica, estudios de imagen del tórax, y las pruebas de función pulmonar. La biopsia de pulmón no suele ser necesaria a menos que exista incertidumbre diagnóstica a pesar de estos estudios no invasivos2-4-5.
Los anticuerpos anti Jo están relacionados en el 80% de los casos con el compromiso pulmonar en estas patologías. Es importante establecer el diagnóstico temprano, ya que el grado de neumonitis intersticial marca el pronóstico en estos pacientes.
Tres de los cuatro casos tuvieron mala evolución a pesar del tratamiento en dos ellos la complicación infecciosa fue la causa de muerte. Un solo paciente tuvo buena evolución5-6-7.
Todos los pacientes se presentaron con características clínicas similares en relación al compromiso pulmonar, afectación poco frecuente (40 - 10%) en pacientes con miopatías inflamatorias. Se resalta la instalación aguda y la gravedad del cuadro con el que se presentaron estos pacientes, enfatizando la negatividad del anticuerpo anti-Jo en los 4 casos, no acorde con la literatura. Referimos estos casos a fin de resaltar la importancia de la investigación de la afectación pulmonar, que en nuestros pacientes marcó el pronóstico.
REFERENCIAS
1- Dalakas MC Signaling patways andinmunology of inflammatory myopathies.Nat clinprt rheumatol 2 :219,2006 [ Links ]
2- Marie I. Polimyositis. And dermatommyositis:Short term and long outcome and predictive factors of prognosis.JRheumatol 28:2230,2008 [ Links ]
3- A. Movasat Hajkhan, A.I. Sánchez Atrio, A. Pérez Gómez y C. Bohórquez Heras. Protocolo diagnóstico y de tratamiento de la hipertensión pulmonar en enfermedades reumáticas. Medicine. 2009;10(32):2173-5 [ Links ]
4- Robert W. Hallowell, Dana P. Ascherman, Sonye K.Pulmonary Manifestations of Polymyositis/Dermatomyositis Semin Respir Crit Care Med 2014; 35(02): 239-248. [ Links ]
5- Dellaripa P, Miller M Interstitial lung disease in dermatomyositis and polymyositis: Clinical manifestations and diagnosis. Uptodate: Jan 13, 2014. [ Links ]
6- Yuko Matsuki, Hiroyuki Yamashita , Yuko Takahashi, Toshikazu Kano, Arisa Shimizu , Kenji Itoh. Diffuse alveolar damage in patients with dermatomyositis: a six-case series Modern RheumatologyApril 2012, Vol. 22, No. 2, Pages 243-248. [ Links ]
7- Dobloug C, Garen T, Bitter H. Ann Rheum Dis. Prevalence and clinical characteristics of adult polymyositis and dermatomyositis; data from a large and unselected Norwegian cohort. 2014 Apr 2. doi: 10.1136/annrheumdis-2013-205127. [Epub ahead of print]. [ Links ]
8- Le Goff B, Chérin P, Cantagrel A, Gayraud M, Hachulla E. Pneumomediastinum in interstitial lung disease associated with dermatomyositis and polymyositis. Arthritis Rheum. 2009 Jan 15;61(1):108-18. doi: 10.1002/art.24372. [ Links ]
9- Imbert-Masseau A, Hamidou M, Agard C, GRolleau JY, Chérin P. Antisynthetase Syndrome. Joint Bone Spine. 2003;70(3):161-168. [ Links ]
10- Sato S, Hirataka M, Kuwana M. Autoantibodies to a 140 KD polypeptides, CDAM 140, in japanese patient with clinically amyipathic dermatomyositis. Arth Rheum 2005;52:1571-1576. [ Links ]
11- Targoff IN. Autoantibodies and their significance in myositis. Current Rheumatology Rep 2008; 10:333-340. [ Links ]
12- Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H, Devulder B, Courtois H. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol. 2001 Oct;28(10):2230-7. [ Links ]
13- Airio A, Kautiainen H, Hakala M. Prognosis and mortality of polymyositis and dermatomyositis patients. Clin Rheumatol. 2006 Mar;25(2):234-9. Epub 2006 Feb 14. [ Links ]
14- Fathi M, Lundberg IE, Tornling G. Pulmonary complications of polymyositis and dermatomyositis. Semin Respir Crit Care Med. 2007 Aug;28(4):451-8. [ Links ]
15- Fathi M, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol. 2005 Nov;17(6):701-6. [ Links ]
16- Miguel Mesa, Luis Fernando Pinto, Carlos Jaime Velásquez. Enfermedad pulmonar vol. 17 No. 4 - 2010. Revista Colombiana de Reumatología Vol. 17 Nº. 4, Diciembre 2010, pp. 257-264 © 2010, Asociación Colombiana de Reumatología. [ Links ]
1. Departamento de Medicina Interna. Servicio de Clínica Médica. Hospital Nacional. Ministerio de Salud pública y Bienestar Social (Itauguá, Paraguay)
Correo electrónico: e.tboggino@hotmail.com
Artículo recibido: mayo de 2014. Artículo Aprobado: 26 de junio de 2014

