









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
El término vasculitis se refiere a la inflamación y necrosis de los vasos sanguíneos, independientemente del tipo de vaso, etiología del proceso u órgano afecto 1,2. Es de etiología no bien conocida, y en general, producida por un mecanismo inmunológico, cuyo órgano diana es el endotelio vascular 1-4. El cuadro puede ser idiopático o secundario a infecciones, fármacos, neoplasias o enfermedades inflamatorias sistémicas 1-3. Puede verse como única manifestación de la enfermedad, siendo esta una vasculitis cutánea localizada 1,4, o en el contexto de cuadros con compromiso sistémico, constituyendo una vasculitis sistémica con compromiso cutáneo. Estos síndromes vasculíticos son infrecuentes 5.
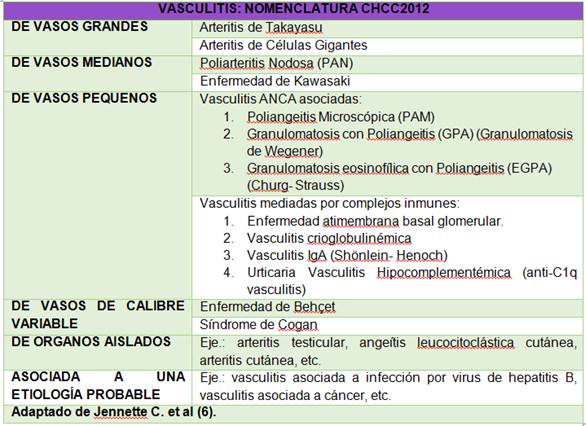
La clasificación de las vasculitis ha sido un tema confuso y de debate en el último medio siglo. En el año 1990 el Colegio Americano de Reumatología publicó una clasificación en función de diversos criterios clínicos e histopatológicos 1. En un intento por mejorar la clasificación Carlson et al. 1,6, proponen un esquema de trabajo dirigido a clasificar las vasculitis cutáneas teniendo en cuenta, el calibre del vaso afecto y el tipo celular predominante 6. Podría decirse que actualmente la clasificación más aceptada para las vasculitis es la nomenclatura de la Conferencia de Consenso de Chapel-Hill del año 2012, que integra los conocimientos sobre la etiología, patogénesis, patología, la demografía y las manifestaciones clínicas 4,7 (Tabla 1).
La afectación cutánea se puede presentar en los distintos síndromes vasculíticos. Los hallazgos clínicos de las vasculitis son variados e incluyen máculas, pápulas, púrpura, lívedo, petequias, úlcera, nódulos y necrosis 1-3 (Figura 1, Figura 2 y Figura 3). El tamaño del vaso afecto se correlaciona con la clínica. Las úlceras, nódulos, cicatrices, lívedo reticularis y necrosis se asocian con la afectación de los vasos arteriales-musculares, y orientan hacia una vasculitis persistente y recurrente, y con presencia de enfermedad sistémica 6,8.
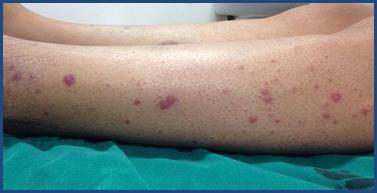
Figura 1 Clínica. Pápulas y placas eritematovioláceas de bordes regulares y límites netos en pierna de paciente con GPA.
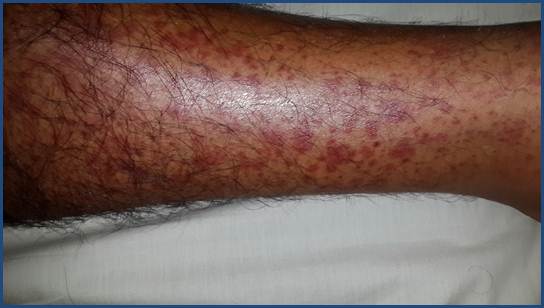
Figura 2 Clínica. Pápulas eritematovioláceas (púrpura palpable) y petequias en pierna de paciente con PSH. Iconografía Cátedra de Dermatología FCM-UNA.
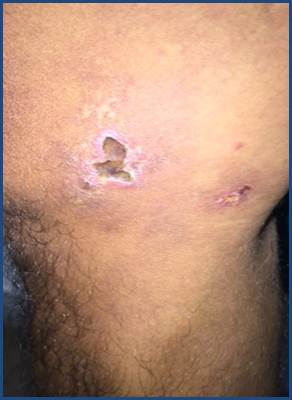
Figura 3 Clínica. Úlceras ovaladas y estrelladas, de entre 2 y 4cm, cubiertas de costras serohemáticas, rodeadas de halo eritematoso, bordes irregulares, límites netos, que asientan cara interna de la rodilla derecha. Paciente con PAM. Iconografía Cátedra de Dermatología FCM-UNA.
El diagnóstico de vasculitis sistémica se establece en pacientes con afectación múltiple y variada de órganos, demostrada por la clínica, los estudios laboratoriales y/o radiológicos 3,5,8 y por la histopatología que evidencia el proceso inflamatorio en la pared del vaso 3,5 (Figura 4). En las vasculitis con afectación sistémica los estudios de imágenes pueden valorar la extensión de la enfermedad, y los estudios serológicos, tales como proteína C reactiva o anticuerpos anti citoplasma de neutrófilos (ANCA) permiten valorar la actividad y el riesgo de morbimortalidad respectivamente 3.
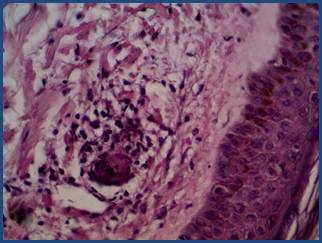
Figura 4 Histopatología. Vaso de pequeño calibre de la dermis superficial con presencia de neutrófilos intraparietales, leucocitoclasia y fibrina intravascular (HE40X). Cortesía Prof. Dra. Beatriz Di Martino.
El tratamiento requiere corticosteroides e incluso el uso de agentes inmunosupresores 5.
MATERIALES Y METODOS
Se realizó un estudio descriptivo, observacional, retrospectivo, de corte transversal, con componente analítico. La población enfocada fueron pacientes atendidos en la Cátedra de Dermatología del Hospital de Clínicas FCM-UNA, ya sea de manera ambulatoria en consultorio o por medio de una interconsulta, durante el periodo de enero de 2004 a diciembre de 2015. Se incluyeron pacientes de ambos sexos y de cualquier edad con diagnóstico clínico de vasculitis sistémica con compromiso cutáneo, y con confirmación histopatológica por biopsia de piel.
El muestreo fue no probabilístico de casos consecutivos, obteniéndose 11 casos de vasculitis sistémicas con compromiso cutáneo.
Se confeccionó una planilla con las variables a consignar. Mediante revisión de historias clínicas y de registros histopatológicos se obtuvo la información de estas variables que comprendieron: Sexo, edad, tiempo de evolución, procedencia del caso (consultorio cátedra de dermatología o interconsultas de otros servicios), lesión dermatológica elemental, síntomas acompañantes, localización de las lesiones, presencia de colagenopatías, marcadores serológicos para colagenopatías y ANCA, características histopatológicas, diagnóstico y tratamiento instaurado.
La gestión y análisis de datos se realizó en una planilla electrónica (Excel del programa Microsoft Office® 2013) y posteriormente se elaboraron tablas y gráficos con estadística descriptiva.
Se aplicó el principio de respeto, beneficencia y privacidad resguardando la identidad de todos los pacientes.
RESULTADOS
De los 112.616 pacientes atendidos de enero de 2004 a diciembre 2015, período de 12 años, 127 correspondían a vasculitis lo que representa una frecuencia global de 0,11 %. De entre estos 127 casos, 11 correspondían a vasculitis sistémica con compromiso cutáneo, lo que corresponde al 8,66% de las consultas por vasculitis.
En el grupo de vasculitis sistémicas con compromiso cutáneo, se encontró un predominio del sexo masculino con el 63,63% de los casos (7/11), frente al 36,36% (4/11) del femenino. Las edades estaban comprendidas entre 6 y 58 años; siendo la edad media 30,18; la mediana 32 y la moda 34,5 años. Para medir la distribución por edades se dividieron en décadas, resultando el rango más afecto el constituido de 31 a 40 años con el 36,36% (4/11). De los 11 pacientes, el 18,18% (2/11) eran niños, siendo todos del sexo masculino.
El tiempo de evolución de las lesiones oscilaba entre dos días y cinco meses. Para su análisis se dividió en semanas, siendo lo más frecuente la consulta a la semana de evolución con el 36,36% (4/11).
En cuanto a la procedencia de las consultas, el 63,63% (7/11) correspondía a pacientes de interconsultas, y el 36,36% (4/11) a pacientes del consultorio propiamente.
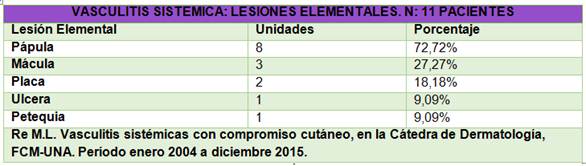
La lesión elemental más frecuente fue la pápula eritematoviolácea (púrpura), que se presentó en el 72,72% (8/11) de los casos. Existen pacientes que han presentado más de un tipo de lesión elemental (Tabla 2). Predominó la localización de las lesiones en miembros inferiores con el 72,72% (8/11) de los casos. La úlcera fue presentada en un caso adulto con PAM, las placas y máculas eritematovioláceas en casos adultos con PAM y GPA. Las pápulas se han presentado en el paciente con GPA y en pacientes con PSH, tanto adultos como niños, mientras que las petequias en un caso de PSH infantil. Predominó la localización de las lesiones exclusivamente en miembros inferiores con el 72,72% (8/11) de los casos.
En la totalidad de los casos se presentaron síntomas, siendo los más frecuentes el dolor abdominal y la artralgia con el 63,63% (7/11) y el 54,54% (6/11) respectivamente.
Se constató afectación gastrointestinal, en el 63,63% (7/11) de los casos, evidenciado por dolor abdominal tipo cólico (7 casos) y/o rectorragia (dos casos); renal en el 36,36% (4/11), evidenciado por hematuria (tres casos), y/o proteinuria (un caso con Proteinuria de 24 hs de 656 mg/24 horas para un valor normal de ˂150mg/24 horas); y pulmonar en el 18,18% (2/11), evidenciado por imágenes de tomografía axial computarizada de alta resolución del tórax (un caso con imágenes nodulares compatible con GPA, y un caso con infiltrado alveolar y vidrio esmerilado compatible con PAM). Cabe resaltar que existieron pacientes con afectación de más de un órgano.
En el 18,18% de los casos (2/11, un caso de Artritis Reumatoide y un caso de Síndrome de Superposición.), se constató concomitancia con colagenopatías.
En lo que respecta a marcadores serológicos para colagenopatías, la batería solicitada ha sido variable; por lo tanto, se ha considerado como perfil reumatológico básico al constituido por los anticuerpos ANA, Anti DNA, Factor Reumatoide; y complemento sérico C3 y C4. ANCAc y ANCAp sólo se ha podido solicitar en dos casos. En el 27,27% (3/11; 2 Factor Reumatoide y 1 ANCA c) de los casos se presentó algún marcador positivo, en el 63,63% fueron negativos (7/11) y en el 9,09%, correspondiente a un caso del total (1/11), el paciente no ha vuelto a la consulta con los estudios solicitados.
En cuanto a la clasificación de las vasculitis según el tamaño del vaso afecto, el 81,81% de los casos (9/11) correspondía a vasculitis de vasos pequeños, mientras que el 18,18% (2/11), a vasculitis de vasos predominantemente pequeños y medianos. En lo que respecta a extensión de la afectación, el 72,72% del total (8/11) presentaba afectación perivascular superficial, mientras que el 27,27% (3/11) presentaban afectación de tipo pandérmico y subcutánea. Respecto al componente inflamatorio predominante, en el 72,72% de los casos (8/11) estaba constituido por neutrófilos, mientras que en el 27,27% (3/11) por linfocitos; cabe destacar que éstos últimos eran casos de vasculitis en fase resolutiva.
Atendiendo a los hallazgos clínicos, laboratoriales, histopatológicos, e imagenológicos, hemos encontrado que el 72,72% de los casos (8/11) correspondía a PSH, el 18,18% (2/11) a PAM, y el 9,09% (1/11) a GPA (Granulomatosis de Wegener).
La Prednisona sola fue el tratamiento de elección en el 72,72% de los casos (8/11); además se realizaron antiinflamatorios no esteroideos (AINES) y reposo, bolos de Metilprednisolona y Prednisona con Ciclofosfamida, cada uno con un caso.
DISCUSION
En este trabajo de investigación, que abarca un período de 12 años en la Cátedra de Dermatología FCM - UNA, se ha encontrado que el 0,11% del total de consultas corresponden a vasculitis en general; y que dentro de este grupo, las vasculitis sistémicas con compromiso cutáneo representan el 8,66% de las consultas; lo que concuerda con la literatura y las publicaciones internacionales (3, 8-10) de que es una patología de baja incidencia y prevalencia. En el estudio realizado por Ochoa D., y col. 10, sobre la epidemiología de las vasculitis primarias en Colombia, se refiere una incidencia promedio de 0,3 a 20 casos por millón de habitantes.
Gota y Mandell 5, refieren una incidencia anual global de entre 10 y 42 casos por millón de habitantes. Por su parte Zazueta B., y Flores L. 8, refieren que estas vasculitis plantean grandes retos a los epidemiólogos, puesto que son raras y se necesita una gran población para determinar la incidencia y la prevalencia. En nuestro país no existen estudios similares que abarquen las características epidemiológicas, clínicas e histopatológicas sobre esta patología, por lo que este trabajo responde a la necesidad de generar datos a nivel nacional y regional inclusive.
Respecto a la distribución por sexo, hallamos predominio masculino lo que se correlaciona con la literatura respecto a que la PAM, la PSH y la GPA, que son las vasculitis más frecuentemente halladas en este estudio, tienen un predominio masculino 1,8.
El rango etario más afectado en este estudio fue la 4ª década de la vida dato no coincidente con lo mencionado en la publicación de Zazueta B., y Flores L. 8, donde se describe una edad pico de aparición de 65 a 74 años. Esto podría deberse a que nuestro hospital se encuentra en el departamento Central (área urbana), el cual presenta un 60% de población entre los 15 y 64 años de edad 11.
El 18,18% eran niños, siendo todos del sexo masculino; lo que va acorde a lo referido por Ting T. (1, 2), en su publicación sobre vasculitis cutáneas en la infancia, de que puede haber una predilección por el sexo masculino; además refiere que debido a la rareza de los casos infantiles de vasculitis cutánea, la información sobre su incidencia y prevalencia es limitada. En el trabajo de Gardner J. y col. 13, realizado en el Reino Unido en el año 2002, que muestra una incidencia anual global estimada de vasculitis primaria en los niños de 20,4/100.000, la PSH es la más prevalente, coincidente con este trabajo, donde los dos casos infantiles se correspondían con PSH.
El tiempo de evolución de las lesiones oscilaba entre dos días y cinco meses, siendo lo más frecuente la consulta a los siete días, hecho que podría deberse a la estoicidad de nuestra población y al mal hábito de la consulta tardía. Es sabido que es imprescindible realizar la biopsia dentro de las 48 horas, y de una lesión representativa 3,5.
El 63,63% de los casos eran provenientes de interconsultas de otros servicios principalmente de clínica médica, lo que evidencia la necesidad de un manejo interdisciplinario y la jerarquización del diagnóstico dermatopatológico.
La lesión elemental más frecuente fue la pápula eritematoviolácea (púrpura palpable) con el 72,72% de los casos, lo que concuerda con los hallazgos de López D., y col. 14, y de Villavicencio A. y col. 15, quienes encontraron la púrpura palpable como lesión elemental más frecuente, con el 62% y el 33,9% de sus casos respectivamente.
En la totalidad de los casos se presentaron síntomas, siendo los más frecuentes el dolor abdominal y la artralgia, lo que concuerda con la literatura de que las vasculitis sistémicas se acompañan de síntomas generales muchas veces inespecíficos 1,7,8.
En nuestro estudio, la localización predominante de las lesiones fue en miembros inferiores con 72,72% de los casos; tal como lo refieren los artículos de Pulido A. y col. 1, Marzano A. 2, Carlson J. y col. 3, y los resultados de los estudios de López D. y col. 14, Villavicencio A. y col. 15, y con el 93,7% y 52,5% respectivamente. Esto es debido a que todas estas manifestaciones son más frecuentes en los miembros inferiores, probablemente por causas hemodinámicas, al menos en el paciente que puede deambular.
Se constató afectación gastrointestinal en el 63,63% de los casos, renal en el 36,36% y pulmonar en el 18,18%, lo que se explica por el hecho de que la mayoría de nuestros pacientes presentaron diagnóstico de PSH, seguido de PAM y luego de GPA. Según lo descrito en la literatura en aproximadamente dos tercios de los pacientes con PSH existe afectación gastrointestinal (16); en la PAM, los principales órganos comprometidos son el riñón (79%-90%) y el pulmón (25%-50%) 1; y en la GPA en el 80% de los casos puede presentarse afectación renal durante la evolución de la enfermedad, generalmente posterior a manifestaciones respiratorias, y hasta en el 17% de los casos es la manifestación inicial 8.
Los ANCA han demostrado ser de gran utilidad y un biomarcador sensible y específico de uno de los subgrupos más frecuentes de vasculitis sistémica primaria 8. Se ha informado su positividad en el 40% al 95% de los pacientes con GPA, el 40% al 90% de los sujetos con PAM y en el 10% al 70% de los pacientes con Síndrome De Churg-Strauss (SCS) 8. En nuestro estudio probablemente por limitaciones económicas, sólo se ha podido realizar en dos pacientes, siendo positivo ANCA c, en el paciente con GPA.
Incluso en el contexto de la revisión de la nomenclatura de CHCC2012 7, se define a la Vasculitis asociada a ANCA (VAA), como una vasculitis necrotizante, con pocos o ningún depósito inmune, que afecta predominantemente a los vasos pequeños, asociada con ANCA específica para mieloperoxidasa (MPO-ANCA o ANCAp) o proteinasa 3 (PR3-ANCA o ANCAc), debiéndose añadir un prefijo al nombre para indicar reactividad ANCA, es decir, MPOANCA, PR3-ANCA o ANCA-negativo. De esta manera se contempla la posibilidad de una VAA con resultados negativos en las pruebas serológicas para ANCA, pero que de otro modo cumplen con la definición de una VAA 7. Las VAA comprenden la granulomatosis con poliangeítis (Wegener) (GPA), la poliangeítis microscópica (PAM) y el síndrome de Churg-Strauss (SCS) 7,8.
La GPA se caracteriza por el compromiso de las vías respiratorias superiores, inferiores y el glomérulo renal, secundario a la presencia de una vasculitis sistémica acompañada de focos inflamatorios granulomatosos y necróticos; a nivel cutáneo pueden presentarse pápulas en miembros inferiores y úlceras mucocutáneas 1,8.
La PAM es una vasculitis que afecta a vasos de pequeño y mediano calibre con afectación sistémica. Los principales órganos comprometidos son el riñón y el pulmón, en forma de glomerulonefritis rápidamente progresiva y hemorragias pulmonar 1,8. A nivel cutáneo las lesiones son indistinguibles de las que se presentan en las vasculitis cutáneas de pequeño vaso 1.
En el contexto de la ruta diagnóstica de las vasculitis, es conveniente solicitar anticuerpos anti nucleares (ANA), complemento y factor reumatoide, a fin de descartar vasculitis secundaria 8. En nuestro estudio hemos hallado solo dos casos con Factor Reumatoide positivo, que se correspondían con paciente con diagnóstico previo de Artritis Reumatoide (AR) y Síndrome de Superposición, quienes no presentaban signos positivos de actividad.
El patrón histopatológico más frecuente en nuestra población fue el de una vasculitis neutrofílica de vaso de pequeño calibre con extensión perivascular superficial, lo que concuerda, con lo descrito por Carlson J., y Chen K. 6, de que el 60% de los pacientes con vasculitis cutánea tendrá vasculitis neutrofílica de vasos pequeños restringido a la dermis superior.
La clasificación de las vasculitis según la nomenclatura CHCC20127 pareciera ser una de las más completas y actuales aplicables a esta patologías; no obstante debido a limitaciones de recursos, no es factible aplicarla a este trabajo retrospectivo, puesto que inclusive en la actualidad el hallazgo histopatológico de depósitos de IgA en las biopsias, ni la determinación serológica de ANCA son factibles en nuestro hospital escuela.
No obstante en un esfuerzo por clasificar los casos, y apoyándonos en la bibliografía que evidencia distintas clasificaciones aún vigentes 4,8,17, en este trabajo, hemos encontramos ocho casos compatibles con PSH, de los cuales dos eran niños y seis adultos (de entre 19 y 58 años); dos casos compatibles con PAM y un caso compatible con GPA.
Según los criterios para PSH del año 2010, de la European League Against Rheumatism/Paediatric Rheumatology International Trials Organization/Paediatric Rheumatology European Society (EULAR/PRINTO/PRES) 17, los dos pacientes pediátricos de este estudio se corresponden con el diagnóstico de PSH. Estos criterios incluyen púrpura palpable como criterio obligatorio, junto con al menos uno de los siguientes hallazgos: dolor abdominal difuso, vasculitis leucocitoclástica con depósitos predominantes de IgA en la biopsia cutánea, artritis aguda o artralgias en cualquier articulación, y afectación renal evidenciado por proteinuria y/o hematuria 17.
El Colegio Americano de Reumatología, en el año 1990 4,8,17, propuso la presencia de al menos dos de los siguientes cuatro criterios, para el diagnóstico de PSH: púrpura palpable, dolor abdominal agudo, biopsia mostrando granulocitos en las paredes de pequeñas arteriolas o vénulas, o edad ≤ 20 años al comienzo de la enfermedad.
Nuestros seis pacientes adultos con diagnóstico compatible con PSH presentan púrpura palpable en miembros inferiores, dolor abdominal agudo y biopsia cutánea mostrando vasculitis de vasos pequeños con polimorfonucleares (además de artralgia y/o hematuria y/o rectorragia en cuatro de los seis casos).
Respecto al mayor caso de pacientes adultos con PSH en este estudio, cabe resaltar que este es un estudio hecho a partir de datos de los pacientes registrados en la cátedra de dermatología exclusivamente (la cual por varios años estuvo separada físicamente, inclusive en ciudades distintas, de la cátedra de pediatría, la cual tenía un profesional dermatólogo en su propio servicio, por lo que es probable que la mayoría de los probables casos de PSH infantiles no hayan llegado a nuestro servicio), además en nuestro estudio existe una mayor proporción de población adulta (nueve casos) que infantil (dos casos).
En los pacientes con PAM y GPA, el diagnóstico se ha planteado en base a los hallazgos clínicos, la histopatología, la imagen tomográfica, y los laboratorios, compatibles con tales diagnósticos.
Resulta difícil contrastar nuestros hallazgos con los de la literatura, puesto que existen limitados trabajos que describen la epidemiología y la clínica de las vasculitis sistémicas con manifestaciones cutáneas de manera global; la mayoría de los mismos se centran en las vasculitis sistémicas primarias asociadas a ANCA por un lado; o constituyen trabajos de investigación sobre algunas de las entidades individualmente. Por otra parte nuestro trabajo presenta un número limitado de casos como para hacer mayores inferencias al respecto. No obstante, en las publicaciones se refiere que la PSH en la infancia, tiene una incidencia anual de 3 a 26 por 100.000 niños, mientras que en adultos, la enfermedad es rara, con una incidencia anual de 0,1 a 1,8 por 100.000 individuos 16. Respecto a la PAM se refiere una incidencia anual de 7,91 por 1000.000 de individuos, y para la GPA 2,95 por 1000.000 (18).
En lo que respecta a tratamientos realizados, se administró Prednisona exclusivamente en el 72,72% de nuestros casos, AINES más reposo en el 9,09%, bolos de Metilprednisolona en el 9,09%, y Prednisona con Ciclofosfamida en el 9,09%. Las publicaciones sobre manejo de vasculitis refieren que si existe compromiso sistémico el tratamiento inicial debe incluir el uso de glucocorticoides a dosis altas y/ o Ciclofosfamida, en pulsos intravenosos o por vía oral, hasta alcanzar la remisión del cuadro 1,5,18,19, tras lo cual, pueden instaurarse pautas de mantenimiento con azatioprina, micofenolato de mofetilo o Metotrexate. El uso de inmunoglobulinas intravenosas o la plasmaféresis resulta útil en casos seleccionados 1.
CONCLUSIONES
Se ha realizado un trabajo observacional de tal forma a caracterizar al paciente con vasculitis sistémica; se plantearon múltiples variables en este estudio, y se han obtenido datos importantes de cada una de ellas, no obstante no todas las variables se han podido recopilar de la totalidad de la muestra. Esto es reflejo, talvez de la falta de un protocolo a seguir en lo que respecta a esta patología, además del factor económico, muchas veces limitante a la hora de realizar estudios laboratoriales y de imágenes ajenos a la disponibilidad de nuestro hospital escuela, que es público.
Dado que las vasculitis sistémicas constituyen una patología de baja incidencia y prevalencia, y a que existen escasos trabajos que hayan estudiado el perfil epidemiológico de las vasculitis, creemos que este trabajo a pesar de sus limitaciones, podría ser considerado como referencia para futuros estudios clínicos y epidemiológicos.