Las Osteogenesis imperfectas: revisión del tema
Osteogenesis imperfecta
Herreros MB1, Franco R1, Ascurra M2
1). Instituto Nacional de Protección a Personas Excepcionales (INPRO).
2). Instituto de Investigaciones en Ciencias de la Salud (IICS).
RESUMEN
]]> Objetivo: Las osteogénesis imperfectas (OI) son un grupo de patologías genéticas hereditarias del tejido conectivo, que se caracterizan por fragilidad ósea y fracturas. Las osteogénesis imperfectas se clasifican en tipos: I, II, III, IV, V y VI. Las de tipo I y IV se subdividen de acuerdo a la presencia o no, de dentina opalescente y la tipo II se divide en tres subgrupos dependiendo de las características radiológicas. La prevalencia estimada de todos los tipos combinados es de 0,5 en 10.000 nacimientos. Las OI son causadas por mutaciones en los dos genes que codifican las cadenas de colágeno tipo 1, COL1A 1, localizado en el cromosoma 17 y COL1A 2, localizado en el cromosoma 7.El tipo de herencia varia de acuerdo a los tipos y subtipos de OI y la severidad puede ser muy variable, y es diferente hasta en una misma familia, desde individuos sin fracturas, hasta pacientes con múltiples fracturas. Algunas características asociadas incluyen; escleróticas azules, dientes opalescentes, hipoacusia, deformidad de huesos largos y columna vertebral e hiperextensibilidad y luxación articular.
Se puede realizar el diagnostico prenatal de las OI por ecografía y si previamente se conoce la mutación, por el estudio de vellosidades coriales o liquido amniótico por biología molecular, que es altamente confiable. Ha habido muchos avances en el diagnostico y el tratamiento de las OI en los últimos años, motivo por el cual, se presenta una revisión de la clínica, clasificación, tipos de herencia, diagnostico, manejo y el tratamiento de la osteogénesis imperfecta.
Palabras claves: osteogénesis imperfecta, fractura, colágeno, mutación.
ABSTRACT
Objective: Osteogenesis imperfecta (OI) is a group of hereditary genetic conditions of the connective tissue characterized by brittle bones and fractures. Osteogenesis imperfecta is classified as type I, II, III, IV, V, or VI. Types I and IV are subdivided by whether or not opalescent dentin is present, while Type II is divided into three subgroups depending on radiological findings. The combined prevalence for all types is 0.5 for every 10,000 live births. OI are caused by mutations in two genes that code the 1 type collagen chain COL1A1, which is located on chromosome 17, and the COL1A2, located on chromosome 7.
The type inherited varies according to type and subtypes of OI, and the severity may also be quite variable, even within a single family, with some individuals experiencing multiple fractures while others have no fractures at all. Some associated characteristics include blue sclerae, opalescent teeth, hypoacusis, deformity of the long bones and spinal column, and hyperextensibility or dislocation of joints.
OI can be diagnosed prenatally by ultrasound, and if the mutation is known in advance, by testing the chorionic villi or amniotic fluid using techniques of molecular biology, which have a high confidence level. There have been many advances in the diagnosis and treatment of OI in recent years, for which reason this review of the clinical characteristics, classifications, types of heredity, diagnosis, management and treatment of osteogenesis imperfecta is presented.
Key words: osteogenesis imperfecta, fracture, collagen, mutation.
]]>INTRODUCCIÓN
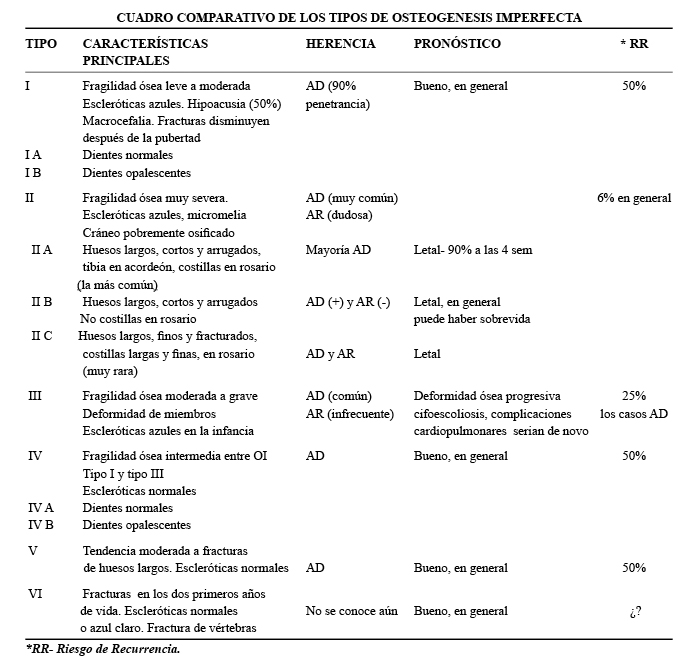
Las osteogénesis imperfectas (OI) son un grupo de patologías genéticas hereditarias del tejido conectivo que se caracterizan por fragilidad ósea y fracturas. Las osteogénesis imperfectas se clasifican en tipos; I, II, III, IV, V y VI (1). Los tipos I y IV se subdividen en grupos I A y IV A, sin dentina opalescente y I B y IV B, con dentina opalescente (1-3).
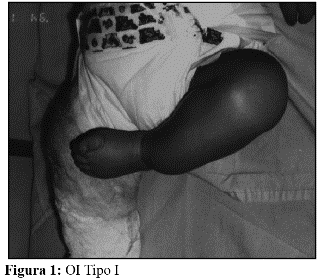
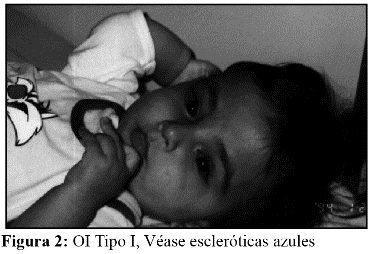
Características clínicas: La OI tipo I es la más leve, presenta fragilidad ósea de leve a moderada, escleróticas azules, macrocefalia y cara triangular. El 8% de los pacientes presenta fracturas al nacimiento y el 23% a lo largo del primer año de vida. En el 35% de los casos se observa hipoacusia. Es de herencia autosómica dominante (AD) y de pronóstico en general, bueno. La severidad de la OI tipo I puede ser muy variable, desde individuos sin fracturas, hasta pacientes con múltiples fracturas. (Fig. 1 y 2).
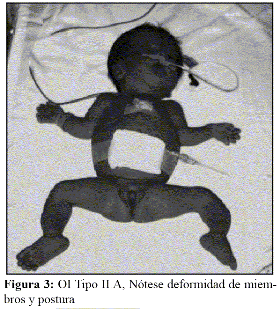
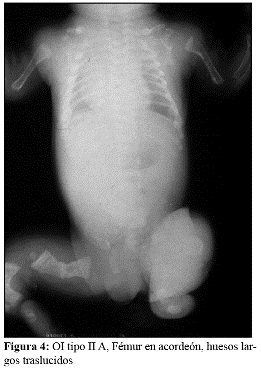

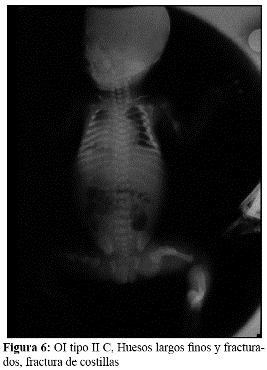
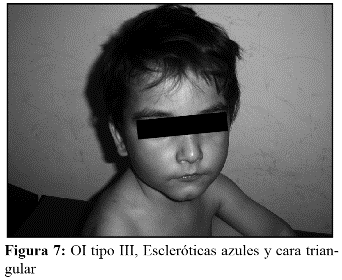
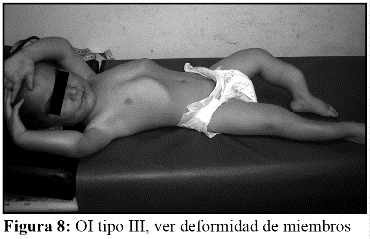
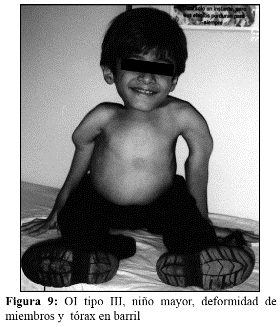
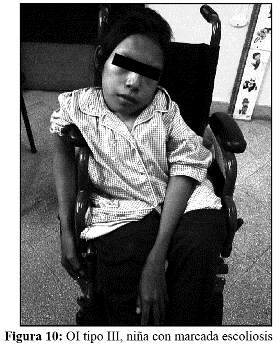
En la OI tipo III, la fragilidad ósea tiene características de moderada a grave con deformidad progresiva de miembros, por lo que los pacientes no desarrollan marcha. Las escleróticas son azules en la infancia, presentan cifoescoliosis, macrocefalia y con frecuencia, dentinogénesis imperfecta e hipoacusia. La OI tipo III es de herencia AD, aunque existe una forma rara AR que es la más común entre los negros sudafricanos. (Fig. 7, 8, 9 y 10).
La OI tipo IV presenta fragilidad ósea intermedia entre la tipo I y la tipo III, las escleróticas son normales, es AD y el pronóstico es bueno, en general (1-7).
En la OI tipo V, se observa una tendencia moderada a la fractura de huesos largos y vértebras, las escleróticas son normales y es AD.
Por ultimo, en la OI tipo VI, se ven fracturas entre los 4 y los 18 meses de vida, escleróticas normales o azul claro, no hay dentinogénesis imperfecta y sí fracturas de vértebras, aun se desconoce el modo de herencia (1).
La habilidad para caminar depende del tipo de OI. Prácticamente en todas las OI tipo I la marcha está conservada, sin embargo las OI tipo III se caracterizan por producir deformidad ósea progresiva y por lo tanto, los pacientes no pueden caminar (1, 2).
]]> La prevalencia estimada de todos los tipos combinados es de 0,5 en 10.000 nacimientos (2).Etiología: las OI son causadas por mutaciones en los dos genes que codifican las cadenas de colágeno tipo 1; COL1A 1, localizado en el cromosoma 17 y COL1A 2, localizado en el cromosoma 7. El tipo de herencia varía de acuerdo a los tipos y subtipos de OI.La OI tipo I, resulta de mutaciones del gen COL1A 1.La OI tipo II se debe a mutaciones del gen COL1A 1 o COL1A 2, la mayoría son mutaciones esporádicas y para estos casos el riesgo de recurrencia es del 6%. En las OI tipo III y IV, se ven mutaciones del gen COL1A 1 o COL1A 2 (1, 2). La tipo V no se asocia a mutaciones del colágeno tipo I y en la tipo VI no se han documentado anomalías de COL1A 1 ni COL1A 2 (1).
Características clínicas asociadas: se describen: escleróticas azules, dientes opalescentes, hipoacusia, deformidad de huesos largos y columna vertebral, hiperextensibilidad articular, retraso del desarrollo motor y talla baja.
El diagnóstico está basado en la historia familiar, características físicas y hallazgos radiológicos (1-8). Se puede realizar el diagnostico prenatal de las OI por ecografía y si se conoce la mutación, con el estudio por biología molecular de vellosidades coriales o liquido amniótico (9).
El diagnóstico diferencial prenatal, es preciso hacerlo con: la Displasia Tanatofórica, la Displasia Campomélica y la Acondrogénesis tipo I; en el neonato con la Hipofosfatasia y en niños mayores con la Osteoporosis juvenil. El diagnostico diferencial mas importante de las OI, I y IV, lo constituye el maltrato infantil (2) (Cuadro Comparativo).
Tratamiento: Las fracturas deben ser diagnosticadas y tratadas lo más tempranamente posible, con un adecuado control de la escoliosis y/o cifosis. El inicio de la terapia física debe realizarse en forma precoz y ésta debe ser personalizada para cada niño. La meta de la terapia de rehabilitación será la de mantener la función óptima en todos los aspectos de la vida del niño. Los padres y cuidadores deben ser detalladamente instruídos acerca del síndrome, y sobre todo de como manejar al paciente, para tratar de disminuir al mínimo el número de fracturas(10). De ser posible, se debería iniciar la marcha en etapa temprana y ésta debe ser estrictamente protegida. Los programas de ejercicios físicos (especialmente la natación), son muy positivos; se recomienda además el uso de colchones especiales para facilitar la movilidad del niño en la cama.
]]> Las fracturas desplazadas deben ser cuidadosamente alineadas y enyesadas y, en caso necesario, se debe recurrir a la cirugía (10).Desde hace algún tiempo se están utilizando los Bifosfonatos para el tratamiento de la OI. Estos son compuestos sintéticos análogos de los pirofosfatos inorgánicos, utilizados con éxito en el tratamiento de la osteoporosis posmenopáusica y de la hipercalcemia del cáncer y actúan inhibiendo la reabsorción ósea por los osteoclastos. Su uso debe ser reservado para casos con síntomas severos en las OI tipo III y IV. Se ha reportado aumento de la densidad ósea, disminución del número de fracturas y del dolor y mejoría en el manejo ambulatorio del paciente. También se reportó aumento del área vertebral.
En general, se utiliza el Pamidronato por vía endovenosa (11- 14), pero existen trabajos que hablan de la eficacia del Alendronato por vía oral, tratamiento que ofrece mayor comodidad de administración y menor costo para el paciente (15).
A nivel neurológico, puede ocurrir una invaginación basilar como complicación, por lo tanto, se impone la realización de tomografías computarizadas de cráneo de control y si fuere necesario, el tratamiento es quirúrgico (2).
Las osteogénesis imperfectas constituyen un grupo de patologías relativamente frecuentes, en algunos casos letales y en otros muy invalidantes; por esta razón es necesario difundir el conocimiento de las mismas, para contribuír a mejorar su diagnóstico, manejo y tratamiento y mitigar en alguna medida, el duro trance que les toca vivir a los niños que la padecen y sus familiares.
REFERENCIAS
1. Jones K. Smith’s Recognizable Patterns Of Human Malformation W.B. 6th Ed. Philadelphia: Elsevier Saunders; 2006.
2. Gorlin Robert J, Cohen M, Hennekam R. C.M. Syndromes Of The Head And The Neck. 4th Edition. New York: Oxford University Press; 2001. [ Links ]
3. Buyse M. Birth Defects Encyclopedia. En: Dover, MA. Center for Birth Defects Information Services.USA: Blackwell Scientific Publications; 1990.p.1321-324. [ Links ]
4. Firth HB, Hurst JA. Oxford Desk Reference - Clinical Genetics. Oxford: Oxford University Press; 2005. [ Links ]
5. Rimoin DL, Connor J, Pyeritz M, Reed E. Emery And Rimoin’s Principles And Practice Of Medical Genetics. 3º Ed. New York: Editorial Churchill Livingstone; 1997.
6. Online Mendelian Inheritance in Man-OMIM [base de datos en Internet]. Maryland: John Hopkins University Press; 2008. [ Links ] Disponible en: http://www. ncbi.nlm.nih.gov/OMIM.
7. Taybi H, Lachman R. Radiology Of Syndromes, Metabolic Disorders And Skeletal Dysplasi.As. 4th edition. Missouri: Mosby; 1996. [ Links ]
8. Vázquez Jesús G, Zafra de la Rosa GF. Atlas: diagnostico De Síndromes Genéticos. Mexico: Manual Moderno; 1999. [ Links ]
9. Jiménez M, Jiménez P, Lozano M, Moro M. Diagnostico Ecográfico de las Displasias Esqueléticas. Prog Diag Prenat. 1998;10(10):555-64. [ Links ]
10. Cassidy Suzanne B, Allanson Judith E. Management Of Genetic Syndromes. 2nd ed. New Jersey: Wiley- Liss; 2005. [ Links ]
11. Pizones J, Plotkin H, Parra-García J, Álvarez P, Gutiérrez P, Bueno A, et al. Bone healing in children with osteogenesis imperfecta treated with bisphosphonates. J Pediatr Orthop. 2005;25(3):332-35. [ Links ]
12. Seikaly M, Kopanati S, Waber P, Patterson D. Impact of alendronate on quality of life in children with osteogenesis imperfecta. J Pediatr Orthop. 2005;25(6):786-91. [ Links ]
13. Vyskocil V, Pikner R, Kutílek S. Effect of alendronate therapy in children with osteogenesis imperfecta. Joint Bone Spine. 2005;72(5):416-23. [ Links ]
14. Di Meglio L, Ford L, McClintock C, Peacock M. Intravenous pamidronate treatment of children under 36 months of age with osteogenesis imperfecta. Bone. 2004;35(5):1038-45. [ Links ]
15. Dimeglio L, Ford L, McClintock C, Peacock M. A comparison of oral and intravenous bisphosphonate therapy for children with osteogenesis imperfecta. J Pediatr Endocrinol Metab. 2005;18(1):43-53. [ Links ] ]]>