Monosomía 18p con características de Síndrome de Kabuki
18p Monosomy with characteristics of Kabuki Syndrome.
Elodia Torres, Maria Beatriz de Herreros, Marta Ascurra.
Departamento de Genética. Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción. Asunción – Paraguay.
Solicitud de sobretiros: Elodia Torres. Dirección: Río de la Plata y Lagerenza – Instituto de Investigaciones en Ciencias de la Salud. Barrio Sajonia. Asunción.
RESUMEN
]]> Niña de 7 años de edad, que consulta por dismorfias faciales y alteraciones del lenguaje, además de otras características clínicas compatibles con el Síndrome de Kabuki.Se realizaron los estudios cromosómicos con técnicas de coloración convencional y de identificación con Bandas G y C, que revelaron una deleción del brazo corto de uno de los cromosomas del par 18.
El cariotipo de la niña resultó 46, XX, del 18(p11.1‡ pter), con lo que se confirmó en la misma el Síndrome 18p-. El cariotipo de los padres fue normal.
Se destaca la importancia del estudio cromosómico en pacientes con características clínicas compatibles con síndromes génicos conocidos, asimismo se compara el fenotipo observado en la paciente con los reportados para el Síndrome de Kabuki.
Palabras claves: deleción 18p, monosomía, Síndrome de Kabuki.
SUMMARY
A 7 year old girl presented with facial dysmorphysms and language disorders, as well as other clinical characteristics consistent with Kabuki Syndrome.
Chromosomal studies with conventional staining techniques and G and C band identification showed a deletion in the short arm of one chromosome 18.
The girl’s karyotype was 46, XX, del 18(p11.1‡ pter), confirming the 18p- syndrome. The parents’ karyotypes were normal.
]]> We highlight the importance of chromosomal studies in patients with clinical characteristics consistent with known genetic syndromes. Furthermore, the patients phenotype is compared with those reported for Kabuki Syndrome.Key words: 18p deletion, monosomy, Kabuki Syndrome.
INTRODUCCIÓN
La deleción del brazo corto del cromosoma 18 fue observada por primera vez por De Grouchy y col., en el año 1963. A partir de entonces han sido reportados más de 100 casos (1). Está considerado como uno de los más frecuentes síndromes de deleción autosómica (2). La relación masculino – femenino es de 3F:2M; en la mayoría de los pacientes reportados la edad promedio de los padres es avanzada. En el 85% de los casos corresponden a deleciones de novo (3) y el resto a productos de un desequilibrio de una translocación balanceada de los padres (4). Típicamente los individuos afectados por la monosomía 18p, tienen retardo del desarrollo psicomotriz, baja estatura por deficiencia de la hormona de crecimiento, dismorfismo facial con cara redonda, orejas prominentes, ptosis palpebral, boca en carpa, severas caries dentales, cuello corto y anomalías esqueléticas (5,6). Aunque no siempre, la extensión de la deleción del brazo corto se correlaciona con las características clínicas del paciente.
El Síndrome de Kabuki, es estimado con una frecuencia de 1 en 32.000 japoneses y 1 en 50.000 caucásicos (7). Denominado así por la similitud con el maquillaje de los actores de teatro Kabuki, una forma de expresión teatral japonesa. Fue descrita en 1981 por Niikawa y col. y Kuroki y col. Desde entonces el número de pacientes ha ido en aumento (8), aún cuando el diagnóstico se complica por la variabilidad clínica que presenta este síndrome. Se lo asocia con una etiología de origen génico, autosómico dominante con expresividad variable; sin embargo ninguna evidencia pudo ser establecida al respecto, como tampoco lo ha sido en relación a algún sitio cromosómico específico. El diagnóstico clínico del Síndrome de Kabuki se basa en cinco elementos principales: rostro peculiar típico, anomalías de los dermatoglifos, anomalías esqueléticas, leve a moderado retardo mental y baja estatura, además de anomalías orales (9-12).
DESCRIPCION DEL CASO CLINICO
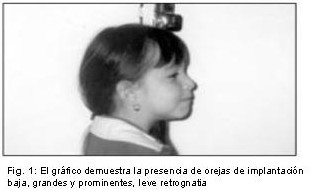
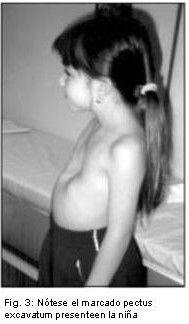
Se reporta el caso de una niña de 7 años de edad, hija de madre de 47 años y padre de 52, no consanguíneos, que consulta por dismorfias faciales y alteraciones del lenguaje. Al examen físico presentó talla de 101 cm. (percentil 3), circunferencia cefálica (CC) de 51 cm. (percentil 50), fisuras palpebrales largas, ptosis moderada, orejas dismórficas, prominentes y de implantación baja, dientes separados y cariados, pectus excavatum, braquidactília y clinodactília de ambos quintos dedos, así como las almohadillas de los dedos algo más gruesas, talla baja y leve retardo mental.
Por las características clínicas, se sospecho que la niña era portadora del Síndrome de Kabuki, y se solicitó un estudio citogenético a fin de descartar una cromosomopatía y /o corroborar el diagnóstico clínico, (Fig. 1,2,3).
]]>
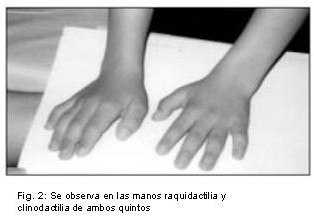
Se sometió a la niña y sus progenitores a estudios cromosómicos, a través del cultivo de linfocitos de sangre periférica, en medio RPMI enriquecido con 15% de suero fetal bovino y fitohemaglutinina (M-form, liofilizado), a 37º durante 72 horas. Transcurrido el tiempo se agregó colchicina (0,003 ug/ml) durante 90 minutos, seguido por tratamiento con solución hipotónica de KCl (0,075M), y fijación en 3:1 de metanol ácido acético. Se realizaron los extendidos sobre portaobjetos, los que se tiñeron con Giemsa para el estudio convencional. La identificación cromosómica se llevó cabo con las técnicas de bandas G y C (13).
]]> Cuarenta células se analizaron por paciente. En todos se encontró, con tinción convencional, 46 cromosomas. Con la técnica de Bandeo G se detectó, en todas las metafases observadas de la niña, la presencia de una deleción del brazo corto de uno de los cromosomas del par 18, región p11.1, (Fig. 4). Ninguna anomalía se observó en los cromosomas de los progenitores descartándose el síndrome de Kabuki.
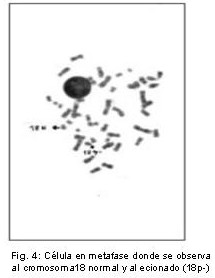
El cariotipo de la niña reveló 46, XX, del (18) (p11.1).
El cariotipo de la madre fue 46,XX y del padre 46,XY.
DISCUSION
Teniendo en cuenta que el cariotipo de los padres fue normal y no habiendo antecedentes familiares que expliquen la presencia de la cromosomopatía, el origen de la misma sería de novo, probablemente en correlación con la edad avanzada de los progenitores (14).
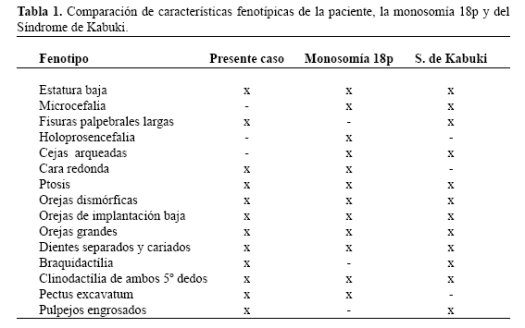
Al examen físico la niña presentaba rasgos de la monosomía para el brazo corto del cromosoma 18, y también del Síndrome de Kabuki (Tabla 1). Para este último, según la Oxford Medical Databases y McKusick, aún no ha sido identificado ningún sitio génico específico, aunque el mismo ha sido relacionado con lugares en los cromosomas autosómicos tales como en el 1p, en el 6q, en el 12q, en el 13q y en los cromosomas sexuales, en el Xp y en el Yp, asociándolo a una herencia autosomica dominante que justificaría la variabilidad clínica presentada por estos pacientes. Este caso sería el primero, reportado en relación al brazo corto del cromosoma 18.
]]> Una vez más se destaca la importancia de la realización del estudio cromosómico, aún en individuos con características fenotípicas compatibles con un síndrome génico conocido, teniendo en cuenta que la presencia de una cromosomopatía permite asociar y de repetirse esta, inferir sobre un sitio génico dado.
BIBLIOGRAFÍA
1. Jones KL, Smith S. Recognizable Patterns of Human Malformation. 5th Edition. Philadelphia.USA. W.B. Saunders Company; 1997. [ Links ]
2. Voiculescu I, Toder R, Back E, Oswald P, Schempp W. A retrospective CISS hybridization analysis of case with de novo translocation t(18;22) resulting in an 18p- Syndrome. Clin Genet. 1993;43:318-20. [ Links ]
3. Rimoin DL, Connor JM, Pyeritz RE. Emery and Rimoin´s Principles and Practice of MEDICAL GENETICS. 3th Edition. USA. Churchill Levingstone; 1997. [ Links ]
4. Gorlin RJ, Cohen M, Michael JR, Hennekam RCM. Syndromes of the Head and the Neck. Oxford University Press. 4th Edition. USA: Oxford University Press; 2001. [ Links ]
5. Artman G, Morris XA, Stock AD. 18p- Syndrome and Hipopituitarism. J Med Genet. 1992;29:671-71. [ Links ]
6. Buyse ML. Birth Defects Encyclopedia: a service of the Center For Birth Defects Information Services. USA: Inc, Blackwell Scientific PUblications; 1990. p. 381-2. [ Links ]
7. Mihci E, Tacoy S, Haspolat S, Karaali K. Central Nervous System Abnormalities in Kabuki (Niikawa- Kuroki) Syndrome. American Journal of Medical Genetics. 2002;111:448-49. [ Links ]
8. McGaughran J, Aftimos S, Jefferies C, Winship I. Clinical phenotypes of nine cases of Kabuki Syndrome from New Zealand. Clinical Dysmorphology. 2001;10:257-62. [ Links ]
9. Matsune K, Shimizu T, Tohma T, Asada Y, OACI H, Maeda T. Craniofacial and Dental Characteristics ofKabuki Syndrome. American Journal of Medical Genetics. 2001;98:185-90. [ Links ]
10. Winter R, Baraitser M. Oxford Medical Databases: Dysmorphology-Kabuki make-up syndrome. Oxford: Oxford University Press. Electronic Publishing; 1996. [ Links ]
11. McKusick VA. On line Mendelian Inheritance in Man. USA: The Johns Hopkins University Press-OMIM; 2003. [ Links ]
12. Asano T, Ikeuchi T, Shinohara R, Enokido H, Hashimoto K. Partial 18q trisomy and 18p monosomy resulting from a maternal pericentric invertion, inv (18)(p11.2; q21.3). Jpn J Human Genet. 1991;36:257-65. [ Links ]
13. Verma RS, Babu A. Human Chromosomes: manual of basic techniques. USA: Pergamon Press; 1989. [ Links ]
14. Thompson T, Mclnnes R, Willard H. Genética en Medicina. 4ª Edición. Barcelona –España: Masson S.A; 1996.
]]>