









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La Enfermedad de Vogt-Koyanagi-Harada (VKH), también denominada síndrome uveomeníngeo, se caracteriza por la afección ocular, donde el hallazgo habitual en la patología establecida es una panuveítis granulomatosa bilateral, que cursa con desprendimiento de retina seroso, edema o hiperemia del disco óptico y puede acompañarse de compromiso del sistema nervioso central, trastornos dermatológicos y auditivos(1). Es una afección de etiología autoinmune y severa, que afecta preferentemente a personas jóvenes y de mediana edad.
La enfermedad de VKH ocurre comúnmente en personas de razas pigmentadas y la teoría más aceptada sobre su origen es la de una reacción autoinmune mediada por células T contra los melanocitos(2). El curso de la enfermedad habitualmente implica cuatros fases: prodrómica, uveítica aguda, convaleciente y crónica recurrente(2,3). Los desencadenantes de esta enfermedad son desconocidos hasta el momento, pero los da tos señalan que en la patogenia existe una importante relación con factores genéticos(4).
La prevalencia de VKH presenta variaciones geográficas, en EE. UU. constituye 1-4% de todas las uveítis y en Japón del 6,7 al 9,2%. Se calcula que el 25% de las personas afectadas por esta enfermedad presentan ceguera legal; 25% quedan con baja visión y sólo 50% con una buena agudeza visual mayor de 20/50(5).
Se han identificado varios factores de riesgo para VKH, incluyen la pigmentación oscura de la piel, las razas oriental, hispana y negra, sexo femenino, edades entre 20 y 50 años. Numerosos artículos han reportado la asociación entre VKH y los genotipos del locus HLA- DRB1(6). La causa exacta de la enfermedad por la que se origina la enfermedad de VKH aún sigue oculta, pero estudios inmunológicos proponen como principal mecanismo un proceso liderado por células T dirigido contra diferentes antígenos asociados a los melanocitos, como la tirosina y otras proteínas afines con la tirosina, que se acoplan al péptido codificado por el gen HLA DRB1*0405(7). Los genes del sistema HLA des- empeñan un papel sumamente importante en el reconocimiento del sistema inmune. El HLA DR es una molécula receptora de la superficie celular que pertenece al Complejo Mayor de Histocompatibilidad (CMH) de clase II y que exhibe los antígenos peptídicos elaborados por el gen HLA DRB1, cuando el sistema inmune no reconoce estos antígenos como propios sino como extraños desencadena la respuesta inmune de rechazo. En los pacientes con VKH se ha reportado que los epítopos de los melanocitos están sensibilizados, y aquellos que poseen HLA DRB1*0405 reconocen un abanico más amplio de péptidos derivados de los melanocitos(6).
La influencia de los factores genéticos en la expresión de VKH, ha quedado en evidencia ante la alta tasa de la enfermedad en determinadas poblaciones como en: japoneses, chinos, indios americanos, hispanos, indios asiáticos y descendientes provenientes de Oriente Medio, asimismo la baja frecuencia en poblaciones caucásicas o de raza negra(1,4,8). Se han investigado po- limorfismos de HLA y otros genes relacionados con la respuesta inmune(4), encontrándose una fuerte asocia- ción con genes HLA de Clase II, como el DR1 y DR4, principalmente con el alelo DRB1*04:05 en diversas poblaciones(4,9).
La enfermedad de VKH clásicamente inicia en la fase prodrómica con síntomas neuroauditivos, seguida de la fase uveítica aguda y por último da origen a las manifestaciones crónicas (oculares y tegumentarias)(10,11). Sin embargo, no es raro que los pacientes se presenten solo con el compromiso ocular, sin los componentes neuroauditivos(11,12). El retraso en el diagnóstico o en la instalación del tratamiento propicia mayores riesgos de cronicidad, complicaciones y discapacidad visual(11-13). Es de suma importancia por ese motivo re- conocer las características distintivas de VKH en la fase temprana del compromiso ocular(11). Existe evidencia de que es de suma importancia diferenciar la etapa en la que se encuentra la enfermedad, dado que una enfermedad ocular de reciente inicio es potencialmente curable, independientemente de que existan o no síntomas neuroauditivos(11).
Los Criterios Diagnósticos de la enfermedad VKH del Comité Internacional de Nomenclatura publicados en el 2001(1), están diseñados para clasificar la enfermedad como Definida o Probable. La categoría Enfermedad Definida se subdivide a su vez en las formas Completa e Incompleta, en la primera se encuentran las manifestaciones oculares, neuroauditivas y tegumentarias, y en la incompleta sumada a las manifestaciones oculares se constatan solo las neuroauditivas o las tegumentarias(1). Son requisitos: la ausencia de antecedente de trauma ocular, el compromiso bilateral y haberse descartado otras patologías diferentes a VKH(1). La limitación que poseen estos criterios revisados es que no ofrecen un diagnóstico definitivo para pacientes sin compromiso extraocular(14).
El diagnóstico de VKH es fundamentalmente clínico; pero en pacientes sin afectaciones extraoculares, la angiografía fluoresceínica y con indocianina, la tomo- grafía de coherencia óptica (OCT), la punción lumbar y la ultrasonografía, pueden ser útiles para confirmarla(5).
En Paraguay no existen registros de la prevalencia de esta afección, es de suma importancia analizar las características clínicas y genéticas de la enfermedad en pacientes de esta población, lo que permitirá establecer protocolos de manejo diagnóstico y terapéutico adecuados. La enfermedad de VKH afecta principalmente a personas en jóvenes, posee mal pronóstico visual si se realiza el diagnóstico y el tratamiento en forma tardía, contar con biomarcadores genéticos susceptibilidad pueden ser de gran ayuda para realizar el diagnóstico temprano o incluso ser útiles para predecir la aparición en determinados grupos de riesgo y tomar las precauciones correspondientes.
METODOLOGÍA
Diseño: Estudio de casos y controles, de susceptibilidad genética para VKH
Población de estudio
Casos: Se incluyeron los pacientes con VKH que recurrieron al consultorio de Enfermedades Oculares Autoinmunes y al consultorio general de enfermedades reumáticas, ambos del Departamento de Reumatología del Hospital de Clínicas (San Lorenzo, Paraguay) por remisión de servicios de oftalmología, desde enero del 2010 hasta julio del 2019.
Los criterios de inclusión fueron: pacientes paraguayos de raza mestiza, mayores de 18 años, con VKH confirmada por los criterios diagnósticos revisados del Comité Internacional de Nomenclatura (Read 2001), que los agrupa en 3 categorías: VKH completo, VKH incompleto y VKH probable, según los componentes clínicos presentes en el paciente.
Registro clínico: de los pacientes con VKH se obtuvieron datos sociodemográficos; etapa en que fue diagnosticada la enfermedad (aguda, convaleciente o crónica); y el espectro de manifestaciones clínicas tanto oculares como extraoculares en cada fase de la enfermedad (prodrómica, aguda, convaleciente y crónica). Se revisó el historial clínico para determinar la presencia de reactantes de fase aguda (proteína C reactiva y velocidad de eritrosedimentación globular); anticuerpos antinucleares (ANA) y factor reumatoide (FR) (marcadores típicos de autoinmunidad); punción lumbar para estudio de LCR con presencia de pleocitosis a expensas de mononucleares. Se registró el tratamiento recibido (corticoides e inmunosupresores) así como las complicaciones oculares (cataratas, glaucoma, sinequias) y la agudeza visual final.
Asociación genética: se realizó en estos pacientes con VKH el genotipado del HLA II DRB1 para determi nar la existencia de susceptibilidad genética la enfermedad.
Controles: Donantes voluntarios de la base de datos del BIOBANCO IMIDs del Paraguay. Se contó con 32 controles sanos, paraguayos de raza mestiza, sin ante- cedentes de enfermedades autoinmunes.
Genotipado HLA DRB1: Se realizó la tipificación de HLA (Human leukocyte antigen) de tipo II haplotipo DR con sus diferentes alelos a los pacientes con VKH y a los controles sanos del BIOBANCO. La tecnología utilizada SSO (oligonucleótidos con especificidad de secuencia) por LUMINEX, que permitió determinar los alelos de HLA DR presentes en una muestra de ADN, amplificada por PCR (lifecodes HLA SSO).
Análisis estadístico: Se realizó con software estadístico SPSS versión 20. La frecuencia alélica se determinó por conteo simple. La prueba exacta de Fisher se utilizó para evaluar las diferencias estadísticas de la distribución de alelos HLA DR B1 entre pacientes y controles, es una prueba de contraste de hipótesis utilizadas en tablas de contingencia. Se calcularon los odds ratios (OR) para los alelos que mostraban diferencias significativas entre la frecuencia en pacientes y en controles; Se definieron intervalos de confianza (IC) del 95% para todas las desviaciones estadística. Un valor p de menos de 0.05 se consideró significativo(15-17).
Aspectos Éticos: Este estudio fue valorado y aprobado por la Dirección de Investigaciones y el Comité de ética de investigaciones de la Facultad de Ciencias Médicas de la Universidad Nacional de Asunción, conforme a lo establecido en el Manual de Normas y Funciones. Resolución Num: 108/2019, del 13 de mayo del 2019. y mantuvo la estricta adhesión a la Declaración de Helsinki para la investigación en seres humanos. Los pacientes incluidos fueron informados sobre los objetivos del estudio y firmaron voluntariamente el con- sentimiento informado.
RESULTADOS
Se incluyeron 21 pacientes para la descripción del espectro clínico y demográfico de la cohorte paragua- ya con VKH. El estudio de asociación genética a través del genotipado HLA DRB1 se realizó en 16 de estos pacientes y se comparó con la de los controles sanos. Se excluyó a 5 pacientes por razones logísticas, como pérdida del seguimiento o procedencia de zonas rurales alejadas. Se contó con 32 controles sanos procedentes del BIOBANCO IMID-PY, 25 (78%) del género femenino y 7 (12%) del masculino, edad de 23 a 68 años, promedio 38,5.
Espectro clínico de la enfermedad de VKH en la cohorte paraguaya
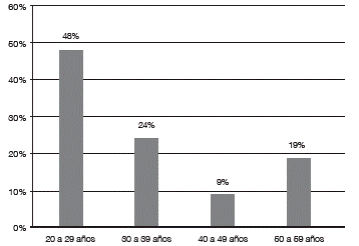
Los pacientes ingresados para el registro clínico de la cohorte de VKH fueron 21, de estos 17 (81%) del sexo femenino y 4 (19%) masculino. La edad de presentación de la enfermedad estuvo en el rango de 20 a 57 años, con un promedio de 35 años, observándose un pico de incidencia en la tercera década (48%), (Gráfico 1).
Siguiendo los criterios diagnósticos del Comité Inter- nacional de Nomenclatura en esta serie de pacientes, predominaron las formas clínicas VKH probable (manifestación sólo ocular) y VKH Incompleto (ocular más otra manifestación neuroauditiva o tegumentaria), sólo un paciente con la forma clínica Completa (Tabla 1 y Tabla 2).
Tabla 1 Clasificación clínica de los pacientes VKH según el compromiso de órganos o sistemas.
| Tipo | Manifestación clínica | Pacientes (%) (n= 21) |
|---|---|---|
| VKH Probable Ocular | -- | 10 (47,6%) |
| VKH Incompleto | Ocular + neuroauditivo o piel | 10 (47,6%) |
| VKH Completo | Ocular + neuroauditivo + piel | 1 (4,7%) |
La etapa evolutiva en que se realizó el diagnóstico de la enfermedad en la mayoría de los pacientes 15 (71%) fue la aguda, específicamente dentro del primer mes de aparición de los síntomas; en 4 casos (19%) en la etapa convaleciente, entre 2 y 4 meses de iniciado el cuadro, y en 2 (10%) pacientes en la etapa crónica, posterior a 4 meses. El tiempo de seguimiento clínico de estos pacientes por el departamento de reumatología fue de 3 a 107 meses, con una media de 37,5 meses.
Tabla 2 Características demográficas, manifestaciones extraoculares y tipo clínico de la cohorte de pacientes con VKH.
| N° | Edad | Género | Auditivos | SNC | Piel | Tipo VKH |
|---|---|---|---|---|---|---|
| 1 | 50 | F | Probable | |||
| 2 | 33 | F | Tinnitus | Pleocitosis en LCR | Incompleto | |
| 3 | 29 | F | Tinnitus | Incompleto | ||
| 4 | 39 | F | Tinnitus | Pleocitosis en LCR | Incompleto | |
| 5 | 20 | F | Tinnitus/hipoacusia | Vitíligo | Completo | |
| 6 | 29 | F | Tinnitus | Incompleto | ||
| 7 | 20 | F | Probable | |||
| 8 | 27 | F | Poliosis | Incompleto | ||
| 9 | 26 | F | Pleocitosis en LCR | Incompleto | ||
| 10 | 38 | F | Tinnitus | Incompleto | ||
| 11 | 37 | F | Probable | |||
| 12 | 52 | F | Tinnitus | Meningismo | Incompleto | |
| 13 | 24 | F | Probable | |||
| 14 | 57 | F | Probable | |||
| 15 | 29 | F | Probable | |||
| 16 | 45 | F | Probable | |||
| 17 | 47 | F | Meningismo | Incompleto | ||
| 18 | 52 | M | Probable | |||
| 19 | 26 | M | Vitíligo | Incompleto | ||
| 20 | 30 | M | Probable | |||
| 21 | 25 | M | Probable |
Las manifestaciones más frecuentes en la etapa prodrómica fueron la cefalea y el dolor periorbitario, seguidos por dolor y congestión ocular, meningismo, tinnitus, náuseas, vómitos, sólo un caso con hipoacusia (Tabla 3).
En la etapa aguda de la enfermedad o fase uveítica, el 100% de los pacientes presentó disminución marcada de la agudeza visual (visión borrosa) de rápida progresión (1 a 4 días), bilateral en 20 (95%) casos y sólo en una paciente fue unilateral al momento del diagnóstico, pero posteriormente al 7mo mes de enfermedad se afectó el ojo contralateral.
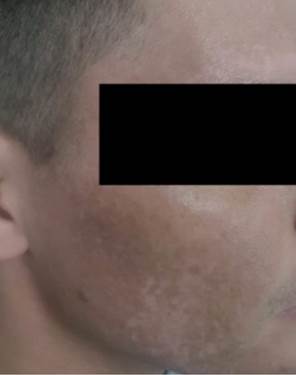
Figura 1. Paciente de la cohorte paraguaya con VKH Tipo Incompleto, a nivel cutáneo con zonas despigmentación en el rostro (vitíligo).
Tabla 3 Manifestaciones clínicas de pacientes con VKH en la etapa prodrómica.
| Síntomas | Pacientes (%) (n=21) |
|---|---|
| Cefalea | 14 (66,6%) |
| Dolor periorbitario | 13 (61,9%) |
| Tinnitus | 7 (33,3%) |
| Dolor y congestión ocular | 6 (28,5%) |
| Meningismo | 4 (19%) |
| Fotofobia | 3 (14,2%) |
| Náuseas y vómitos | 3 (14,2%) |
| Síndrome pseudogripal | 1 (4,7%) |
| Hipoacusia | 1 (4,7%) |
| fosfenos | 1 (4,7%) |
En la fase de convalecencia se produce la des- pigmentación coroidea y los cambios tegumentarios. Compromiso de piel y faneras en 3 (14,3%), 2 casos con vitíligo (Figura 1) y uno con poliosis (Tabla 2). A nivel ocular en la mayoría de los pacientes 15 (71%) se constataron alteraciones pigmentarias del EPR, con formación de las típicas imágenes Sunset glow en el fondo de ojo.
Los hallazgos oftalmológicos en la atapa aguda se especifican en la Tabla 4, en 1 caso no se pudo definir debido a la larga evolución de la enfermedad y el diagnóstico se había realizado en otro centro.
En la analítica sanguínea, se realizó la determinación de anticuerpos antinucleares (ANA) en 16 de los 21 pacientes, fue negativa en 15 (93%) y sólo en 1 caso fue positiva con titulación baja de 1:80; el Factor Reumatoide se buscó en 9 pacientes y fue negativo en todos.
Tabla 4 Hallazgos oftalmológicos en la fase aguda VKH.
| Signos | Pacientes (%) (n= 20) |
|---|---|
| Panuveítis | 19 (95%) |
| - Bilateral | 18 (90%) |
| - Unilateral | 1 (5%) |
| Uveítis Posterior bilateral | 1 (5%) |
| Desprendimiento de retina | 19 (95%) |
| Edema de papila | 5 (25%) |
Tabla 5 Resumen AV final de los pacientes con VKH.
| AV | Número de ojos (%) n=40 |
|---|---|
| 20/20 a 20/50 | 27 (67,5%) |
| 20/50 a 20/200 | 8 (20%) |
| 20/200 o menos | 5 (12,5%) |
los casos. Se realizó la determinación de reactantes de fase aguda en 18 pacientes, la PCR fue positiva en 5 (28%), y la VSG fue superior a 20 mm en 6 casos (33%). La punción lumbar y estudio de LCR se hizo a 12 pacientes, se constató alteración en 4 (33%) casos, consistente en pleocitosis a expensas de mononucleares.
Las secuelas y complicaciones oculares fueron: 6 (28,5%) cataratas, 3 (14%) glaucoma, 5 (24%) sinequias anteriores/posteriores. La mejor Agudeza Visual (AV) final se registró en 20 pacientes, se valora individualmente la visión en 40 ojos (Tabla 5). La mayoría quedó (67,5%) con buena visión, 12,5% presentó ceguera legal que corresponde a 5 ojos, 3 pacientes con afectación unilateral y una paciente con ceguera bilateral.
Todos los pacientes recibieron corticoides como parte del tratamiento. Se administró en forma intravenosa al momento del diagnóstico a 18 (85,7%) pacientes, metilprednisolona 1g/día por 3 días consecutivos en 8 (44%) casos y por 5 días en 10 (55,5%). Los 21 pacientes fueron tratados en el primer mes con prednisona 1mg/kg/d en promedio (dosis de 60 a 75 mg/d), a los 3 meses la dosis promedio de prednisona fue 37 mg/d, a los 6 meses 23 mg/d y a los 12 meses de 10 mg/d.
El tratamiento con inmunosupresores al momento del diagnóstico se realizó en 20 pacientes. La inducción con Ciclofosfamida IV mensual en 8 (40%) y con Azatioprina 12 (60%). De los pacientes tratados con Azatioprina inicialmente, en 5 casos fue necesario cambiar de fármaco y se optó por Ciclofosfamida, los motivos, falta de respuesta o reactivación en 4 casos y en 1 paciente por intolerancia a Azatioprina.
El tratamiento de mantenimiento en 13 (62%) pacientes fue con azatioprina, con MTX en 4 (19%) y asociación de ambos fármacos en 3 (14%). Se presentó un solo caso refractario, que recibió además de los inmunosupresores mencionados previamente, Infliximab con respuesta parcial y finalmente Rituximab lográndose la remisión, actualmente con MTX de mantenimiento.
La fase crónica con patrón de uveítis anterior recurrente se constató en 12 (57%) pacientes, 8 (38%) tuvieron solo 1 episodio y 3 (14%) pacientes 2 episodios, que fueron superados con ajuste de la dosis y ritmo de descenso de corticoides más el ajuste de la dosis del inmunosupresor o cambio de este, y solo 1 (4,7%) caso con múltiples reactivaciones (6 episodios de uveítis) que requirió varios esquemas de tratamiento hasta llegar a medicación biológica.
Análisis de HLA DR B1 en pacientes paraguayos con VKH
Los resultados del genotipado HLA II DR en los casos y controles se muestran en la Tabla 6 y Tabla 7. Se realizó en primera instancia un análisis por grupos de alelos y luego por alelos específicos. Se destaca que los alelos del grupo HLA DRB1*01 estuvieron presentes en 5 (31%) de los 16 pacientes y ausente en los controles sanos, con un valor estadísticamente significativo (p 0,04); HLA DRB1*04 se encontró en 6 (37,5%) de los casos y en 10 (31,2%) de los controles. En 3 (18,7%) pacientes se observó la presencia de ambos grupos de alelos.
En el análisis de los alelos específicos HLA DRB1 de los pacientes y controles, ninguno de estos fue estadísticamente significativo. Se destaca que en los pacientes con VKH el alelo predominante fue DRB1.
Tabla 6 Genotipado: Frecuencia de Grupos de alelos HLA II DR en pacientes y controles sanos.
| HLA II | Pacientes n=16 | Frecuencia (%) | Controles n=32 | Frecuencia (%) | Odds ratio (IC 95%) | p |
|---|---|---|---|---|---|---|
| DRB1*01 | 5 | 31,2 | 0 | 0 | n/a | 0,004 |
| DRB1*03 | 2 | 12,5 | 3 | 9,3 | 1.27 (0.20-8.00) | 0,981 |
| DRB1*04 | 6 | 37,5 | 10 | 31,2 | 1.15 (0.37-3.53) | 0,781 |
| DRB1*07 | 2 | 12,5 | 5 | 16,6 | 0.73 (0.13-4.01) | 0,536 |
| DRB1*08 | 4 | 25 | 16 | 50 | 0.39 (0.12-1.30) | 0,094 |
| DRB1*09 | 1 | 6,2 | 0 | 0 | n/a | 0,348 |
| DRB1*10 | 1 | 6,2 | 1 | 3,1 | 1.90 (0.12-31.48) | 0,577 |
| DRB1*11 | 4 | 25 | 10 | 31,2 | 0.71 (0.21-2.49) | 0,420 |
| DRB1*13 | 1 | 6,2 | 6 | 18,7 | 0.29 (0.03-2.52) | 0,227 |
| DRB1*14 | 5 | 31,2 | 3 | 9,3 | 3.52 (0.78-15.81) | 0,094 |
| DRB1*16 | 1 | 6,2 | 6 | 18,7 | 0.29 (0.03-2.52) | 0,227 |
Tabla 7 Genotipado: Alelos específicos del HLA DR B1 en pacientes con VKH y controles.
| DRB1 | Pacientes n=16 | Frecuencia Alélica (%) | Controles n=32 | Frecuencia Alélica (%) | Odds ratio | Evaluación p |
|---|---|---|---|---|---|---|
| DRB1 01:01 | 1 | 3,1 | 0 | - | ||
| DRB1 01:02 | 4 | 12,5 | 0 | - | ||
| DRB1 03:01 | 2 | 6,3 | 1 | 3,8 | 1,667 | 0,579 |
| DRB1 04:01 | 1 | 3,1 | 1 | 3,8 | 0,806 | 0,700 |
| DRB1 04:04 | 1 | 3,1 | 2 | 7,7 | 0,387 | 0,421 |
| DRB1 04:05 | 1 | 3,1 | 2 | 7,7 | 0,387 | 0,421 |
| DRB1 04:11 | 1 | 3,1 | 3 | 11,5 | 0,247 | 0,231 |
| DRB1 07:01 | 2 | 6,3 | 5 | 19,2 | 0,280 | 0,135 |
| DRB1 08:01 | 1 | 3,1 | 4 | 15,4 | 0,177 | 0,119 |
| DRB1 08:04 | 3 | 9,4 | 1 | 3,8 | 2,586 | 0,389 |
| DRB1 09:01 | 1 | 3,1 | 0 | - | ||
| DRB1 10:01 | 1 | 3,1 | 1 | 3,8 | 0,806 | 0,700 |
| DRB1 11:01 | 1 | 3,1 | 2 | 7,7 | 0,387 | 0,421 |
| DRB1 11:03 | 1 | 3,1 | 0 | - | ||
| DRB1 11:04 | 2 | 6,3 | 0 | - | ||
| DRB1 13:02 | 1 | 3,1 | 2 | 7,7 | 0,387 | 0,421 |
| DRB1 14:01 | 1 | 3,1 | 0 | - | ||
| DRB1 14:02 | 3 | 9,4 | 1 | 3,8 | 2,586 | 0,389 |
| DRB1 14:06 | 1 | 3,1 | 0 | - | ||
| DRB1 16:01 | 1 | 3,1 | 1 | 3,8 | 0,806 | 0,700 |
DISCUSIÓN
Este estudio ha descripto el espectro clínico y el genotipo HLA DRB1 de pacientes paraguayos con VKH, se observó un franco predominio del género femenino, la edad media de debut de la enfermedad fue de 35 años, y en mayor proporción fueron afectadas las personas en la tercera década de vida. En relación a estos datos demográficos existen varias publicaciones, como: Aláez et al, en la serie de casos de mexicanos mestizos con VKH y otra realizada por Chee et al., de Singapur, donde reportaron edad promedio de 41 años, también con predominancia del género femenino(18), Singhal et al., describió en una población hindú los mismos datos con respecto a la edad pero mayoritariamente pacientes del sexo masculino(9); Yang P. et al, desde China, en uno de los estudios con mayor población (410 pacientes), informó que 52% correspondían al género masculino(19).
Según lo informado en las distintas publicaciones, la frecuencia es algo superior en mujeres(18), se puede afirmar que las poblaciones asiáticas reportan una distribución de la enfermedad más equitativa en cuanto al género, en pacientes hispanos esta brecha aumenta y es mucho más frecuente en mujeres, y en la cohorte paraguaya es muy preponderante el género femenino. Con respecto a la edad de presentación, el rango es bastante amplio, pero se constata en la literatura científica que es por lo menos 5 años superior a la encontrada en esta cohorte.
Según los criterios diagnósticos revisados(1), en esta cohorte solo un caso de los 21 cumplió los criterios para ser definido como VKH Completo, en el resto de los pacientes la distribución fue equitativa para las formas de VKH Incompleto (con toque ocular más neuroauditivo o tegumentario) y VKH Probable (solo compromiso ocular).
El componente auditivo se manifestó en un tercio de los pacientes de este estudio, en la etapas prodrómica y aguda, todos estos con tinnitus o acúfenos y solo un caso asociado a hipoacusia. Los síntomas cedieron con el tratamiento y ninguno de los pacientes quedó con secuela auditiva. En la serie de pacientes hispanos del Sur de California, las anormalidades auditivas no alcanzaron al 30%, similar a esta población paragua- ya, sin embargo, en las series japonesas llegan hasta un 75%(8). La afectación neurológica se observó en la cuarta parte de los casos de esta cohorte, las manifestaciones fueron meningismo y pleocitosis aislada en LCR. La despigmentación en piel y faneras tuvo una muy baja frecuencia en esta población, solo en 3 de los 21 pacientes, y ningún caso con alopecia.
En los pacientes hindúes los síntomas más frecuentes fueron cefalea y disminución de AV, la afectación auditiva fue muy baja solo 7% en comparación a la nuestra(9). En mexicanos la enfermedad es clínicamente similar a la reportado en otras series, pero la alopecia y la cefalea se observan con mayor frecuencia(4). En el norte de China, las manifestaciones extraoculares se observaron en 88% de 634 pacientes, siendo más frecuentes la afectación neurológica, seguida de poliosis, tinnitus, alopecia y vitíligo(14).
Las alteraciones pigmentarias a nivel del EPR son un equivalente de las lesiones hipopigmentarias de la piel, corresponden a las alteraciones de la fase convaleciente conocidas como "Sunset glow" en el fondo de ojo y estas se observaron en la mayoría de los pacientes paraguayos.
Con respecto a la agudeza visual, la mayoría de los pacientes de esta cohorte quedó con una buena visión (20/20 a 20/50) luego de resolverse el proceso inflamatorio, esto probablemente esté relacionado con el diagnóstico precoz y el tratamiento agresivo de inicio con bolos intravenosos de corticoides y la asociación de inmunosupresores. La peor visión, es decir ceguera legal se produjo en 5 de 40 ojos correspondientes a 4 pacientes, dos de ellos diagnosticados y tratados en la etapa crónica. Concha del Río et al., en un estudio con 26 pacientes encontró la AV mayor a 20/40 en el 75% de los pacientes, tanto del grupo de corticoides como el de inmunosupresores(20).
Todos los pacientes paraguayos recibieron corticoides sistémicos, la mayoría en forma de bolo IV de Metilprednisolona 1g/día entre 3 a 5 días al momento del diagnóstico y posteriormente prednisona por vía oral a dosis altas con descenso gradual, llegando a 23 mg/ día en promedio a los 6 meses y 10 mg/día al año, cabe destacar que este aumento estadístico del promedio de la dosis de prednisona fue debido a que algunos pacientes sufrieron reactivaciones en los primeros meses de tratamiento y por ende tuvieron ajustes de la medicación.
La fase crónica de la enfermedad caracterizada por la uveítis anterior recurrente se constató en 12 pacientes de esta serie (57%), pero en la mayoría se trató de un episodio único, que no recidivó posterior al reajuste de la dosis de corticoides y de los inmunosupresores, 3 pacientes presentaron un segundo episodio y solo en un caso las reactivaciones fueron múltiples. Una paciente con diagnóstico VKH tipo completo fue la que presentó múltiples episodios de uveítis anterior y fue refractaria al tratamiento por lo cual recibió en forma consecutiva azatioprina, ciclofosfamida, metotrexato, infliximab y rituximab.
Ha quedado demostrado en varios estudios que el retraso en el diagnóstico y en la realización del tratamiento empeora el pronóstico e induce mayores riesgos de evolución a la cronicidad, con eso a las complicaciones y discapacidad visual(11-13). Una terapia agresiva y precoz evitaría el desarrollo de las formas completas de la enfermedad(9).
La influencia de los factores genéticos en la patogénesis de VKH ha sido descrita desde hace unas décadas, las publicaciones sobre el tema señalan principalmente al sistema HLA de clase II con el haplotipo DR. La relación del antígeno HLA DR4 con la enfermedad de VKH ya se había mencionado desde la década de los ochenta por Ohno et al., en población japonesa y posteriormente en China(21).
El genotipado HLA DRB1 en esta cohorte paraguaya no ha identificado de manera estadísticamente significativa ningún alelo específico que esté asociado a la enfermedad de VKH con respecto a los controles sanos, sin embargo, teniendo en cuenta la cantidad de estudios previos que señalan una relación de la enfermedad con determinados grupos de alelos como los HLA-DRB1*01 y DRB1*04 y que se corresponden con los serotipos HLA DR1/DR4, se han analizado también los grupos de alelos más prevalentes.
Los grupos de alelos más frecuentes en los pacientes paraguayos fueron HLA-DRB1*01 y HLA-DRB1*04. Los alelos del grupo HLA-DRB1*01 estaban presentes en un tercio de los pacientes y ausentes en los controles sanos; se ha demostrado que existe dependencia estadísticamente significativa (con una p 0,04) entre el estado del paciente y la pertenencia al DRB1*01, por lo que se puede inferir con estos resultados que los alelos del grupo HLA-DRB1*01 predisponen a la enfermedad. Los alelos del grupo HLA-DRB1*04 estaban presentes en poco más de un tercio de los casos, pero también en la misma proporción en los controles. Se puede resaltar que casi el 20% de los pacientes poseían la presencia de ambos grupos alelos HLA DR1/DR4. Weisz (1994) había encontrado en hispanos del sur de California una significativa proporción de pacientes con HLA DR4 y DR1, pero incluso esta relación fue más alta para HLA DR1(22).
En el análisis de los alelos específicos HLA DRB1, ninguno de estos resultó estadísticamente significativo, pero llamativamente en los pacientes con VKH el ale- lo predominante fue HLA DRB1*01:02 presente en 4 (25%) de casos y ausente en los controles, los demás alelos tuvieron muy baja frecuencia. Probablemente si la muestra fuera de mayor tamaño podría determinarse con claridad si este alelo es candidato a biomarcador de susceptibilidad.
Shindo et al, indicaron el primer hallazgo de una alta asociación de los alelos DRB1*0405 en japoneses (1994) y la presencia de DRB1*04:10 sólo en los enfermos y su ausencia en los sanos, finalmente todos los pacientes expresaban uno u otro de estos genes(21). En la cohorte paraguaya sólo un caso y un control sano presentaron DRB1*04:05, con respecto al DRB1*04:10 fue negativo en todo el grupo. El paciente que expresaba este gen se correspondía con la forma clínica VKH Completa.
Arellanes-García et al, determinaron una asociación más fuerte con HLA-DR4 y sólo un ligero aumento de DR1 en pacientes mexicanos, este aumento de DR1 se interpretó como un epítopo compartido que predispone a la enfermedad(23). En Brasil, Goldberg et al.(7) con 37 pacientes con VKH, demostraron que la mayoría de los pacientes poseían DR4 (54,1%), a su vez varios pacientes presentaban HLA-DR1 en ausencia de DR4. Levinson et al(24), estudiaron los alelos DRB1 y DQB1 en 29 pacientes mestizos del sur de California e identificaron la asociación de HLA-DRB1*04 con VKH, confiriéndole un riesgo moderado, además constataron mayor frecuencia de combinación de los alelos HLA-DRB1*01 y HLA-DRB1*04.
Los resultados de un metaanálisis realizado por Shi et al., indican que los portadores de HLA-DR4 / HLA- DRB1*04 presentan riesgo aumentado de desarrollar VKH. El nivel de esta asociación es superior en el este de Asia y más baja en los hindúes. En relación a los alelos del grupo HLA-DRB1*04, específicamente HLA-DRB1*04:04, 04:05 y 0410 incrementaron el riesgo de VKH; por otro lado 04:01 reduce el riesgo de VKH(6). La mayoría de los trabajos mencionan la relación de VKH con el HLA DR4/HLA-DRB1*04, se observa esto en mayor proporción en asiáticos, las publicaciones que informan asociación del HLA-DR1/HLA-DRB1*01 fueron principalmente en latinoamericanos, lo que coincide con el hallazgo de este trabajo.
Estos resultados incentivan a realizar más estudios en los pacientes con VKH, con una muestra más amplia, el hallazgo de un biomarcador genético de susceptibilidad podría colaborar al diagnóstico precoz, lo cual ha demostrado ser crucial para el pronóstico visual.
CONCLUSIÓN
La enfermedad de VKH en esta cohorte de pacientes paraguayos se presentó en mayor proporción en jóvenes de la tercera década de vida, y del género femenino. Según los criterios diagnósticos revisados las formas clínicas predominantes de VKH fueron los tipos Probable e Incompleto. La agudeza visual final en la mayoría de los pacientes fue buena, en relación con el diagnóstico precoz, el tratamiento con dosis altas de corticoides intravenosos y la asociación de inmunosupresores. Sin embargo, un porcentaje bajo de los pacientes presentó ceguera legal coincidente con el diagnóstico y tratamiento tardío.
Se ha encontrado en esta cohorte paraguaya asociación genética de la enfermedad de VKH con los alelos del grupo HLA DRB1*02, sin embargo, no se halló relación estadísticamente significativa con ningún alelo específico, probablemente esto se debió a que la muestra fue pequeña. En los pacientes con VKH de esta cohorte el alelo predominante fue el HLA DRB1*01:02 presente en 25% de casos y ausente en los controles, podría ser un candidato a biomarcador genético en esta población.
Se requieren estudios con mayor número de pacientes en esta misma línea de trabajo para el hallazgo definitivo de biomarcadores genéticos de susceptibilidad que sean de aplicabilidad en el diagnóstico temprano o de utilidad pronóstica.