









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El Lupus Eritematoso Sistémico (LES) es una enfermedad autoinmune inflamatoria crónica, de causa desconocida, sin especificidad por un órgano en especial1.
El LES Juvenil (LESJ) afecta a un mayor número de órganos y sistemas y presenta un curso clínico más agresivo que en adultos2.
La mejor estimación señala que el LES afecta entre 5.000 y 10.000 niños en los Estados Unidos3. El LES de la niñez afecta a las niñas con más frecuencia que los niños (8:1), incluso en el grupo de edad prepúberes (4:1). Puede ocurrir a cualquier edad, aunque se vuelve más frecuente después de los cinco años de edad y es de vida4.
Los factores patogénicos más invocados son los autoanticuerpos, las células B y T, los factores genéticos, hormonales y ambientales y la apoptosis. La producción de autoanticuerpos es un rasgo característico de los pacientes con LES. Estos anticuerpos pueden ser una pieza clave en la patogenia, una consecuencia del daño tisular o bien la huella de un agente etiológico desconocido5-7.
Se ha descrito una asociación de la enfermedad con antígenos leucocitarios humanos (HLA, por sus siglas en inglés): HLA-DRB1 y HLA-DQB1 se asocian a un riesgo aumentado para LES en población caucásica. HLA-DR2 y HLA-DR3 elevan el riesgo de LES en caucásicos por dos a tres veces8. En latinos con nefritis lúpica se describe mayor prevalencia de HLA-B*08, DRB1*08, y DRB1*159.
En dos estudios retrospectivos de Paraguay, las mujeres adolescentes fueron las más afectadas, los principales motivos de consulta fueron fiebre, dolor articular y lesión en piel, donde además del ANA positivo en todos los pacientes, el siguiente anticuerpo más frecuente fue el anti-DNAdc; y la principal causa de mortalidad fue la infecciosa10-11.
Las manifestaciones hematológicas ocurren en un rango de aproximadamente un 33% a 75% de niños con LES12. La leucopenia se produce en casi dos tercios de los niños en algún momento durante el curso de la enfermedad. Leucopenia se define como un total de glóbulos blancos menor a 4000 / μL. La disminución en el recuento de glóbulos blancos se debe principalmente a una caída en el número absoluto de linfocitos acompañado de un aumento en el porcentaje de granulocitos13. La neutropenia es poco frecuente en el LES, y cuando ocurre, suele asociarse a infección grave, efectos adversos de fármacos, o anticuerpos antineutrófilos12. La anemia se presenta en el 50 a 75 por ciento de los niños afectados. La anemia se define como una concentración de hemoglobina más de dos desviaciones estándar por debajo de la media para la edad y el sexo. Los tipos más comunes de anemia en los niños con LES son anemia de enfermedad crónica, anemia por deficiencia de hierro y anemia hemolítica autoinmune (AHA)12,14. Estos pueden ser vistos por separado o en combinación. Se debe buscar una fuente continua de hemorragia (por ejemplo, pulmones, tracto gastrointestinal) si la prueba directa de antiglobulina es negativa en presencia de un recuento elevado de reticulocitos. La prevalencia reportada de trombocitopenia varía del 7 al 30 por ciento12. La trombocitopenia significativa se define como un recuento de plaquetas de menos de 100.000 / microL. Como regla general, el grado de trombocitopenia es leve, y la hemorragia es rara. Sin embargo, no existe una correlación directa entre el número de plaquetas y la probabilidad de sangrado. En algunos casos, estas diferencias están relacionadas con la gravedad y la actividad de otras manifestaciones del LES o con la terapia15. Varios estudios han demostrado la relación entre el Sx de Evans (anemia hemolítica autoinmune + trombocitopenia) y el LES16-17. Se describe una prevalencia del Síndrome de Evans (AHA más trombocitopenia autoinmune) secundario en LES en un 1.7% a 2.7% de los casos18.
Los criterios de clasificación por la SLICC (Systemic Lupus International Collaborating Clinics) 2012 para el LES son una nueva posterior a los criterios del American College of Rheumatology (ACR) versión 1997, utilizados para adultos19-20. Así, los criterios SLICC tienen una sensibilidad superior a los criterios previos (SLICC 94% vs ACR 86%), pero con una especificidad semejante (ACR 93% y SLICC 92%). Sin embargo, actualmente existen nuevos criterios de clasificación del EULAR/ACR (European League Against Rheumatism/American College of Rheumatology) 201921.
Las alteraciones hematológicas podrían presentarse como única manifestación del LES, por lo que debe realizarse un abordaje sistémico y detallado de dichos pacientes, una vez que se hayan excluido otras causas como infecciones y neoplasias.
El objetivo del presente trabajo fue determinar el compromiso hematológico en pacientes con debut de diagnóstico de LESJ.
MATERIAL Y MÉTODOS
Estudio retrospectivo, observacional, descriptivo, de corte transversal. Muestreo no probabilística de casos consecutivos de LESJ, diagnosticados o en seguimiento durante el periodo de enero de 2012 a mayo de 2017, en la Cátedra y Servicio de Pediatría de la Facultad de Ciencias Médicas de la Universidad Nacional de Asunción.
Se utilizaron criterios de clasificación para LES por SLICC 2012.
RESULTADOS
De los 73 pacientes, se hallaron 57 mujeres (78%) y 16 (22%) varones, con una relación F:M de 3,5:1. La edad promedio al diagnóstico fue de 11,9 años (DE: 3,65, rango entre 2 y 17 años). Procedencia: Departamento Central y Capital 39 pacientes (53%), y del Interior 34 casos (46%).
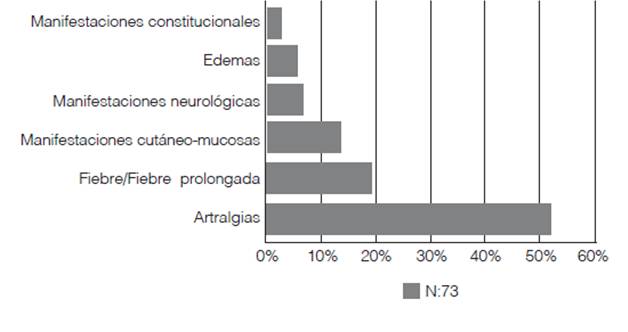
La media del tiempo de evolución desde el inicio de los síntomas fue de 2 meses (DE 1,27 meses). Los síntomas a la primera consulta fueron: artralgias en 38 pacientes (52%), fiebre 14 casos (19,2%), manifestaciones cutáneo-mucosas en 10 pacientes (13,7%), manifestaciones neurológicas en 5 casos (6,8%), edemas en 4 pacientes (5,5%) y manifestaciones constitucionales en 2 pacientes (2,7%). (Figura 1)
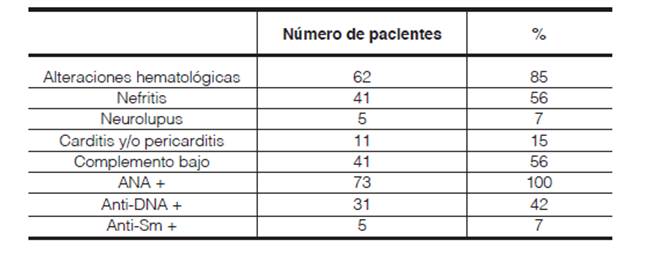
En cuanto a los criterios de clasificación, las alteraciones hematológicas se hallaron en el 85% de los pacientes, toque renal en el 56%, cardiovascular en el 15% y neurológico en el 7% de los casos. (Tabla 1)
Se hallaron alteraciones hematológicas en 62 pacientes (85%), siendo una sola serie afectada en 25 casos (40,3%), bicitopenia en 33 pacientes (53,2%), y pancitopenia en 4 pacientes (6.5%). Las alteraciones más frecuentes fueron: anemia en 58 pacientes (79.4%), linfopenia en 27 casos (36,9%), trombocitopenia en 21 pacientes (28,4%) y leucopenia en 17 casos (23,2%) (Ver Figura 2). El 60% de los pacientes tenían más de una serie afecta. De los pacientes con anemia (n: 58): 16 pacientes (27,6%) presentaron Test de Coombs directo positivo, 42 pacientes (72,4%) Test de Coombs directo negativo. En cuanto a los índices hematimétricos, 40 pacientes (69%) presentaron anemia normocítica normocrómica y 18 pacientes (31%) anemia microcítica hipocrómica. Hallamos Síndrome de Evans en tres pacientes.

DISCUSIÓN
Con respecto al sexo más comprometido existe predominio franco en el sexo femenino, con una relación femenino/masculino 3,5/1, que se asemeja a la relación encontrada por Pluchinotta y cols22 en Italia, sin embargo en el trabajo de Gomes et al23 en Brasil se encontró una relación de 6/1.
En lo que respecta a la edad de presentación, el promedio de 11,9 años coincide con la literatura23-24.
El LES es raro antes de los 5 años, en nuestra serie se presenta un caso de un niño de 2 años. Un 27,3% debutó antes de los 10 años, como se describe por otros autores23-24.
El tiempo de evolución entre el inicio de los síntomas y el diagnóstico es variable, entre 1 mes a 5 años12. En el presente trabajo, el promedio de tiempo de evolución fue de 2 meses. Entre los signos y síntomas más frecuentes hallados se encuentran artralgias, fiebre y manifestaciones cutáneo-mucosas, al igual que en trabajos descritos por Ambrose et al24 y Chiang et al25.
Las manifestaciones hematológicas fueron muy frecuentes (85%), similar a los datos referidos por otros autores23-26.
En esta serie se constató a la anemia como afectación hematológica más frecuente, al igual que en otros trabajos12,25. El 69% de los pacientes con anemia presentaban anemia normocítica normocrómica y un 27,6% anemia hemolítica con test de coombs directo positivo, este último más frecuente que en otros estudios donde lo describen entre 15 y 20% de los casos24,27.
En el estudio realizado por Acosta-Colmán28 y cols en pacientes adultos con LES de nuestro país, describen que puede encontrarse anemia secundaria a enfermedades crónicas, por ferropenia y la hemolítica autoinmune.
La anemia de enfermedades crónicas es la más comúnmente identificada en los pacientes con LES. La anemia ferropénica es la segunda causa de anemia en el LES y puede ser secundaria a perdidas gastrointestinales a metrorragia. La gravedad de la misma generalmente se correlaciona con el grado de la actividad de la enfermedad y con un nivel elevado de eritropoyetina12.
La Leucopenia se observó en 23,2% de los pacientes, inferior a lo reportado por otros autores 35-40%12,29.
La leucopenia se asocia indirectamente con mayor actividad de la enfermedad y con otras manifestaciones como úlceras orales, infecciones y serositis, además de ser un factor predisponente para padecer enfermedades infecciosas que pueden ser mortales. Se con-stató linfopenia en 36,9%, superior a otros estudios, tanto con población caucásica como latina29-30.
La Trombocitopenia se encontró en 28,4%, concordante con lo observado por Gokce et al y Ambrose y cols15,24, pero menor a Gomes et al23. La trombocitopenia también se ha asociado indirectamente con mayor actividad de la enfermedad y, por lo tanto, con tendencia a presentar mayor daño orgánico.
En una cohorte francesa de Síndrome de Evans secundario, se encontró otra enfermedad subyacente en el 10% de los casos, donde se observaron diversas manifestaciones inmunes asociadas (principalmente linfoproliferativas, otras enfermedades autoinmunes e hipogammaglobulinemia) en el 60% de los casos31.
En el presente estudio encontramos 3 pacientes con Síndrome de Evans (4%), muy por debajo de la descripción en otras estadísticas31-32. En nuestra serie se constató pancitopenia en 6,5%, la cual se presenta muy raramente en casos de LES1,33.
Las limitaciones del presente trabajo corresponden al carácter retrospectivo y los factores socioeconómicos insuficientes.
CONCLUSIÓN
Las alteraciones hematológicas fueron un hallazgo frecuente al debut del LES, con afectación de dos series en la mayoría de los casos, siendo la anemia no hemolítica el hallazgo predominante. Las pruebas de laboratorio son de gran valor cuando se evalúa a un paciente con sospecha de enfermedad autoinmune, ya que los resultados pueden confirmar el diagnóstico, estimar la severidad de la enfermedad, evaluar el pronóstico y realizar el seguimiento de la actividad del LES.