











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El término vasculitis se refiere a varias enfermedades que involucran inflamación de los vasos sanguíneos con posterior destrucción de tejido y/o insuficiencia orgánica1. Las vasculitis sistémicas son enfermedades complejas, heterogéneas y potencialmente mortales, objeto de considerable investigación básica y clínica2-3.
Tabla 1 Criterios finales de clasificación para Poliarteritis nodosa infantil (EULAR / PRINTO / PRES).
| Histopatología: Evidencia de vasculitis necrotizante en arterias de mediano o pequeño tamaño. Anomalías angiográficas: Angiografía que demuestra aneurismas, estenosis u oclusión de arterias de mediano o pequeño tamaño, no debido a Displasia fibromuscular u otras causas no inflamatorias. La angiografía convencional es la modalidad de imagen preferida. Histopatología o anomalías angiográficas (obligatorio), además de uno de los cinco criterios siguientes: | |
| Compromiso cutáneo | Livedo reticularis: patrón reticular violáceo por lo general distribuido irregularmente el tejido subcutáneo, a menudo más importante con enfriamiento. Nódulos en la piel: nódulos subcutáneos sensibles. Infartos superficiales de la piel: úlceras superficiales de la piel (piel y tejido celular subcutáneo) u otros cambios isquémicos menores (infartos del lecho ungueal, hemorragias o necrosis digitales). Infartos profundos de la piel: úlceras profundas de la piel (tejido celular subcutáneo y estructuras más profundas); Necrosis/gangrena de falange digital u otros tejidos periféricos (nariz y orejas). |
| Mialgia o miositis | Dolor muscular o inflamación muscular |
| Hipertensión | Presión arterial sistólica/diastólica mayor que el percentil 95 para la estatura. |
| Neuropatía periférica | Neuropatía periférica sensorial: neuropatía con pérdida de sensibilidad con distribución en guantes o calcetines. Meuritis motora: mono/múltiple neuritis motora de nervios periféricos. |
| Compromiso renal | Proteinuria: >0,3gr/24hs, o >30mmol/mg de albúmina en orina/creatinina en orina en una muestra de la mañana. Hematuria o Cilindros hemáticos: > 5 hematíes/campo de alta potencia o cilindros hemáticos en el sedimento urinario o ≥ 2 + en tira reactiva. Deterioro de la función renal: Tasa de Filtrado Glomerular (fórmula de Schwartz) <50% de lo normal. |
Las vasculitis sistémicas son trastornos multisistémicos de los vasos sanguíneos, definidos por el tamaño del vaso que afectan predominantemente, es decir, el tamaño de los vasos pequeños, medianos o grandes, o variable; puntos últimos en los que han basado tradicionalmente su clasificación1,2.
La etiología de todos los trastornos de vasculitis es esquiva. La vasculitis sistémica se divide en dos categorías principales: síndrome de vasculitis primaria y síndrome de vasculitis secundaria. El primero es un tipo de vasculitis causada por la inflamación de los vasos sanguíneos, mientras que el último es inducido por las afecciones subyacentes, que incluyen la enfermedad del tejido conectivo, los tumores, la infección y la alergia a medicamentos4. Las características clínico-patológicas de la vasculitis secundaria y su respuesta terapéutica pueden ser morfológicamente indistinguibles de las de las vasculitis sistémicas primarias y están determinadas en gran medida por el sitio, el tamaño, la extensión y la naturaleza de los vasos sanguíneos afectados5.
Las vasculitis de arterias de tamaño mediano incluyen la poliarteritis nodosa (PAN), PAN cutánea (PANc) y la enfermedad de Kawasaki. Parece probable que los procesos inmunológicos involucrados sean similares a los de otras vasculitis sistémicas e incluyan complejos inmunes, complementos, posiblemente autoanticuerpos, moléculas de adhesión celular, citoquinas, factores de crecimiento, quimiocinas, neutrófilos y células T6.
En 1990, el Colegio Americano de Reumatología (ACR) propuso criterios de clasificación para pacientes con vasculitis en adultos. Para cumplir con los criterios ACR para PAN se requieren la presencia de al menos 3 criterios sobre 10 en total (sensibilidad 82.2%, especificidad 86.6%)7-8.
Por ende, la Conferencia de Consenso Internacional de Chapel Hill (CHCC) de 1994 y 2012, redefinió y estandarizó la nomenclatura de diferentes tipos de vasculitis, además de aclarar las definiciones de enfermedades (diferencias entre PAN y Poliangitis microscópica), cambios hacia el uso de descripciones fisiopatológicas en lugar de epónimos e inclusión de otros tipos de vasculitis como vasculitis de un solo órgano, vasculitis asociada con enfermedad sistémica o asociadas a una causa subyacente8,9. Sin embargo, el CHCC, es un sistema de nomenclatura y no un sistema de clasificación, representando un consenso de expertos en lugar de estar basado en datos1,9. Así, definen a la PAN como la arteritis necrotizante de arterias de mediano y pequeño calibre, sin glomerulonefritis o vasculitis en arteriolas, capilares o vénulas, y sin asociación a anticuerpos anticitoplasma de neutrófilos (ANCA)8.
Los criterios de clasificación ACR de 1990 para las vasculitis sistémicas no incluían niños ni adolescentes, motivo por el cual en 2005, el grupo de trabajo de vasculitis de la Sociedad Europea de Reumatología Pediátrica (PRES) propuso nuevos criterios de clasificación, respaldados por la Liga Europea contra el Reumatismo (EULAR), basados en una revisión de la literatura y un consenso de expertos, validados posteriormente con los mismos y la Organización Internacional de Ensayos de Reumatología Pediátrica (PRINTO), que culminó en la Conferencia de Consenso de Ankara de 2008(>10, >11. Los nuevos criterios de clasificación para Poliarteritis Nodosa infantil se describen en la Tabla 1 10-12.
Si bien el diagnóstico se basa en datos clínicos, son necesarios estudios anatomopatológicos o de imagen para demostrar la presencia de vasculitis.
La PAN es una vasculitis poco frecuente, por lo que presentamos dos casos en niños, uno como PAN sistémica y otro como PANc.
Caso clínico 1:
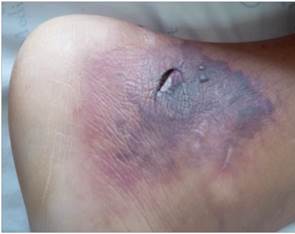
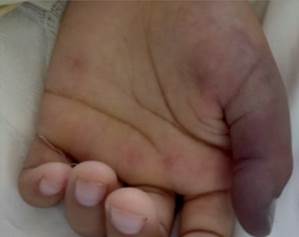
Masculino de 13 años de edad, previamente sano, acude por historia de una semana de evolución de fiebre, artralgias y mialgias, además de edema de miembros inferiores, más lesiones petequiales generalizadas. Ingresa delicado, taquicárdico, taquipneico, pálido, afebril, con mucosas secas, con lesiones equimóticas generalizadas, edema (Godet ++) en MMII izquierdo, tenso, normotérmico, pulso pedio izquierdo disminuido (Figura 1). Se plantea sepsis de foco cutáneo e inicia amplia cobertura antibiótica, además se sospecha de trombosis Venosa Profunda, con EcoDoppler de MMII que informa presencia de signos indirectos de trombosis Venosa Profunda en poplíteo y gemelar izquierdo por lo que inicia Heparina de Bajo Peso Molecular. Al 6to día de internación presenta isquemia a nivel de dedo pulgar izquierdo (Figura 2) por lo que inicia tratamiento con Pentoxifilina. Se realiza laboratorio GB: 12.990/mm3, N: 80%, Plt.: 256.000, Hb: 9,6gr%, Hto: 29%, PCR: + 96mg/L, VSG: 80mm, TP: 65%, INR: 1,65, TTPA: 32, Fibrinógeno: 565, Dimero D menor a 5, GPT: 159, GOT: 126, FA: 514, CK total: 1.378, LDH: 775, Aldolasa: 19,00 (VR hasta 7.5), Alb: 2,9, urea 30, creatinina 0.8, orina simple normal, ASTO neg, ANA: negativo, AntiDNA: negativo, C3: 242 normal, C4: 40.7 normal, perfil inmunológico normal, Hepatitis A, B y C negativos; Biopsia de lesiones en piel: informa Vasculitis Leucocitoclástica + Trombosis en arteriolas. PAMO normal; Por tanto, se cataloga como un caso de Poliarteritis nodosa infantil en base a: 1. Oclusión de arterias medianas y biopsia con vasculitis necrotizante, 2. Compromiso cutáneo (infartos), 3. Miositis. Se inició tratamiento con corticoides, IGIV y ciclofosfamida, con mejoría clínica.
Caso clínico 2:
Masculino de 9 años de edad, previamente sano, con cuadro de fiebre, dolor abdominal, náuseas y vómitos de 4 días de evolución, además de lesiones en piel de tipo nódulos palpables y petequias en miembros inferiores y superiores. Ingresa taquicárdico, con artritis en tobillos, livedo reticularis en miembros inferiores y lesiones descritas anteriormente. Laboratorio GB: 22.000/mm3, N: 89%, linfo 6%, Plt.: 200.000, Hb: 11,7gr%, Hto: 33%, PCR: + 102mg/L, VSG: 40mm, TP: 9%, INR: 1,13, TTPA: 36 seg, Fibrinógeno: 795, GPT: 39, GOT: 39, FA: 180, CK total: 38, LDH: 419, ASTO neg, ANA: negativo, AntiDNA: negativo, ANCA-c neg, ANCA-p neg, C3: 150 normal, C4: 21 normal, HepatitisA, B y C negativos, orina simple normal. Biopsia de lesiones en piel: informa Vasculitis Leucocitoclástica en arterias de pequeño calibre, sin Trombosis y sin depósitos de IgA. Diagnóstico: Poliarteritis nodosa cutánea en base a: 1. Livedo reticularis y nódulos en piel. 2.Vasculitis leucocitoclástica en arterias de pequeño calibre. Se inició tratamiento solamente con corticoides, con excelente respuesta.
DISCUSIÓN
La PAN tiene una incidencia de 4-16 casos por millón, y una prevalencia 31 casos por millón13,14. Se presenta con mayor frecuencia en población asiática15. Los casos de PAN asociados a hepatitis B son menos del 5% en países desarrollados15. Tanto la PAN sistémica como la cutánea son más frecuentes en adultos. Datos de incidencia y prevalencia en niños son más escasos debido a la rareza de la enfermedad en esta edad15. El pico de incidencia en niños es entre 9-10 años17, y en los adultos 40-50 años15. En general afecta a ambos sexos por igual19. La mortalidad global de la PAN en la niñez oscila entre 0-43% debido generalmente a afecta- ción gastrointestinal17. Otras causas de mortalidad son insuficiencia renal, isquemia cerebral. La mortalidad es mayor en el adulto que en el niño14 y sin tratamiento la enfermedad tiene mal pronóstico. No contamos con registros a nivel nacional.
Las vasculitis sistémicas son entidades con características clínicas pleomórficas, y la PAN no está excen- ta de dicha definición. Se caracteriza por un síndrome vasculítico debido a oclusión vascular de diferentes órganos: riñón, sistema nervioso central y periférico, aparato digestivo, miocardio, músculo, vísceras y piel, pudiendo variar de una forma en apariencia limitada a una falla visceral fulminante. Generalmente no afecta al pulmón20. Se sospecha PAN en un paciente con fiebre persistente, pérdida de peso, dolores músculoesqueléticos, sin causa sistémica aparente; isquemia inexplicable en el sistema nervioso central o corazón, datos de abdomen agudo; hipertensión arterial con presencia de sedimento urinario (no causa glomerulonefritis); neuropatía periférica (hipoacusia neurosensorial, mononeuritis múltiple son frecuentes) y afección cutánea; la orquitis es el síntoma más característico de la enfermedad3,13,13,20. En el primer caso descrito, encontramos fiebre prolongada, lesiones en piel, isquemia en dedo con posterior amputación, y miositis; sin embargo, en el segundo caso, lo más llamativo fue la afección cutánea.
La frecuencia de manifestación en diferentes órganos y sistemas depende de su presentación en niños o adultos. Algunos estudios sugieren que la PAN juvenil presenta un curso más benigno, dado por mayor afectación cutánea y renal, que la PAN de inicio en edad adulta, con mayor compromiso neurológico, del sistema gastrointestinal y cardiovascular14,19,21. Además de la forma idiopática sistémica (PAN idiopática generalizada), existen otras 2 variantes clínicas aceptadas, la PAN cutánea y la PAN asociada al virus de la hepatitis B (VHB); variantes clínicas que conllevan implicaciones terapéuticas específicas. La PAN asociado al VHB produce más frecuentemente neuropatía periférica e hipertensión y menos frecuentemente lesiones cutáneas22. En nuestros casos, no hallamos relación con virus de hepatitis, y en ambos casos fue muy importante el compromiso de la piel.
De ser posible debe realizarse de los sitios sintomáticos y optar por los menos invasivos. En la histopatología de la PAN se puede observar: necrosis fibrinoide de arterias de mediano y pequeño calibre con una repuesta inflamatoria marcada alrededor de los vasos, compatible con el caso 1. La histología de la PAN cutánea revela la presencia de vasculitis leucocitoclástica en las arterias de pequeño y mediano tamaño de la dermis profunda e hipodermis, también compatible con el caso 26,7,16,20.
La angiografía visceral está indicada cuando no se puede obtener biopsia, y cuando existe hay compromiso renal, abdominal o cardíaco. Se pueden encontrar microaneurismas saculares o fusiformes, lesiones estenóticas16. También se pueden utilizar técnicas menos invasivas, pero menos específicas como la angio tomografía o angio resonancia.
Antes de iniciar el tratamiento es muy importante definir la severidad de la enfermedad, casos leves: que impliquen síntomas constitucionales, artritis, anemia, lesiones en piel pueden tratarse en principio solo con corticoides y casos moderados a severos con compromiso renal, neurológico, gastrointestinal o isquemia de extremidades es necesario asociar otro inmunosupresor, generalmente ciclofosfamida23.
En PAN asociada a virus de hepatitis b o c debe considerarse el tratamiento antiviral, y si hay manifestaciones graves de vasculitis se podrían utilizar ciclos cortos de inmunosupresores o recambio plasmático24.
En los casos de PAN cutánea asociada a infección por estreptococo beta hemolítico del grupo A se recomienda profilaxis prolongada con penicilina25.
En el manejo de infarto de extremidades, primero deben descartarse otras entidades como infecciones, otras vasculitis ANCA +, y luego iniciar dosis altas de corticoesteroides, vasodilatadores como bloqueantes de canales de calcio, nitroglicerina tópica, etc. En cuanto al uso de sildenafil y pentoxifilina existe menos evidencia con relación a su uso en los niños y más bien en adultos. Para el tratamiento de la hipertensión en la PAN se utilizan los inhibidores de la enzima convertidora de angiotensina (IECA) pero se recomienda monitorizar los niveles de creatinina y si se presentara un aumento reemplazar los IECA por antagonista de canales de calcio. La mayoría de las PANc responden al tratamiento con corticoides pero en el curso de la enfermedad suelen aparecer recaída. Cuando la situación crítica de isquemia está controlada continuar con corticoides vía oral a dosis bajas y otro inmunosupresor como: azatioprina, micofenolato mofetil, dapsona, metotrexato, o hidroxicloroquina por al menos 18 meses3,4,5,15,13. Considerar además uso de dosis bajas en aspirina y heparina de bajo peso. Cuando la necrosis ya está instalada está indicada la cirugía para desbridamiento y amputación del tejido gangrenoso con el fin de evitar la sobreinfección, en algunos casos ocurre la amputación espontánea del tejido necrótico26. En el caso descrito con PAN sistémica, utilizamos corticoides, IGIV, ciclofosfamida y anticoagulación, con remisión de la enfermedad. En el paciente de la PANc encontramos excelente respuesta con el uso de corticoesteroides sin otra asociación.
El uso combinado de corticoides más ciclofosfamida demostró mayor eficacia en el tratamiento para la inducción de la remisión en las PAN moderadas a severas19.Guillevin et al. encontraron que el tratamiento con 12 ciclos de ciclofosfamida versus 6 presentó menor riesgo de recaída, mayor sobrevida libre de eventos y sin diferencia en cuanto a mortalidad27. Falcini et al. reportaron casos de PAN refractaria a ciclofosfamida y corticoides que presentaron mejoría notoria con micofenolato15. La inmunoglobulina se reserva para casos agudos y severos. En casos de PAN refractaria se han utilizado los agentes biológicos como rituximab, tocilizumab e infliximab6,28-30.
En conclusión, la PAN es una vasculitis de rara presentación, que debe sospecharse ante la presencia de fiebre persistente, pérdida de peso, dolores músculo esqueléticos, isquemias, abdomen agudo, hipertensión arterial, alteración en el sedimento urinario, neuropatía periférica, orquitis y afección cutánea. La presunción obliga a la realización de estudios de laboratorios ampliados, biopsias e imágenes vasculares, a fin de diagnosticar y tratar tempranamente.

