











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El Síndrome Antifosfolipídico (SAF) es una entidad autoinmune que se caracteriza por la presencia de trombosis (arterial y/o venosa) y/o morbilidad obstétrica (i.e. pérdidas fetales recurrentes, pre-eclampsia y retardo del crecimiento intrauterino) con la presencia de anticuerpos antifosfolipidos (AAF), anticoagulante lúpico (AL), anticardiolipina (AC) y la anti-B2glicoproteína I1,2.
El SAF es de distribución universal más frecuente en mujeres, en edades entre 20 y 40 años y es actualmente reconocido como una causa mayor de hipercoagulabilidad adquirida. Múltiples estudios han demostrado una baja prevalencia de los AAF en individuos normales, siendo el rango entre 1 al 5% y en personas de tercera edad en un promedio de 12% a los 70 años3),4.
El SAF puede manifestarse en pacientes que no tienen clínica ni evidencia analítica de otra enfermedad de base (SAF primario) o asociarse a otras enfermedades autoinmunes (i.e. lupus eritematoso sistémico, artritis reumatoide, polimiositis, dermatomiositis, esclerosis eistémica, entre otras), a infecciones, neoplasias y al consumo de determinados fármacos, denominándose en este caso como SAF secundario1, (5.
El cuadro clínico del SAF se caracteriza por la presencia de trombosis arterial, morbilidad del embarazo (principalmente, pérdidas fetales) y trombocitopenia moderada. La afectación de un solo vaso o las múltiples oclusiones vasculares pueden dar lugar a una amplia variedad de presentaciones. La gravedad de la manifestación clínica es variable, pudiendo ir desde un cuadro clínico leve, hasta formas severas que pueden poner en peligro la vida del paciente3, (5. Debido a esto, se puede manifestar con trombocitopenia, livedo reticularis, lesiones de la válvula cardíaca, anemia hemolítica, epilepsia, úlceras en las piernas, infarto de miocardio y amaurosis fugax, entre otros3,6,7.
El diagnóstico de esta entidad se basa en criterios clínicos y analíticos, que han sido modificados a lo largo del tiempo. En el año 1999, en Sapporo Japón, se redactaron unos criterios preliminares para la identificación del SAF, pero esta clasificación no contemplaba ciertas características clínicas y laboratoriales, tales como Livedo Reticularis o Trombocitopenia, las cuales se ha comprobado que se presentan con más frecuencia en estos pacientes. Estos criterios han sido evaluados y han mostrado tener una sensibilidad de 71% y una especificidad del 98%8. Posteriormente se realizó un consenso en Sydney en el año 2006, con el objetivo de modificar los criterios previos basados en el avance del conocimiento de la fisiopatología de la enfermedad. De esta forma se incluyó a los anticuerpos anti-B2 glicoproteína. Si bien no se agregaron criterios clínicos, si se propuso la presencia de ciertas manifestaciones clínicas como asociadas al SAF como la afectación cardiaca, el livedo reticularis y la trombocitopenia entre otras manifestaciones5,9,10.
Actualmente existen pocos datos acerca de esta entidad en población paraguaya, por lo cual el objetivo de este estudio ha sido el describir las manifestaciones clínicas y analíticas de los pacientes con diagnóstico de SAF en un hospital universitario.
PACIENTES Y MÉTODO
Tipo de estudio: Estudio retrospectivo, de una serie de casos, consecutivos de corte transverso, realizado en pacientes con diagnóstico de egreso de SAF inter- nados en la I, II y III Cátedra de Clínica Médica del Hospital de Clínicas, y pacientes del consultorio externo del Dpto. de Reumatología del Hospital de Clínicas durante el periodo de enero 2001 a junio 2011.
Se incluyó en el estudio a todos los pacientes que cumplieran con los criterios diagnósticos de SAF11.
Metodología: Se registró un gran número de variables clínicas y analíticas (i.e. edad, sexo, procedencia; presencia de trombosis arterial, venosa y de pequeños vasos, asociación con otras patologías de base, manifestaciones clínicas, la presencia de anticuerpos: anticardiolipina, IgG, IgM, TTPA, Anticoagulante lúpico en plasma, VDRL (+) falso, anti B2GP1isotipos IgG y/o IgM así como la presencia de otros anticuerpos). De forma asociada se registró el número de pacientes fallecidos así como el tratamiento recibido.
Estadística: Se utilizó una estadística descriptiva, con distribución de frecuencias, medidas de tendencia central (moda, mediana, media), medidas de dispersión y Desviación estándar, (DE). El análisis de los da- tos se realizó con en la planilla Excel.
RESULTADOS
Se incluyeron a 25 pacientes (n=25) que cumplían con los criterios diagnósticos de SAF, siendo 23 (92%) de sexo femenino con una edad media de 37,8 años. La mayoría de los pacientes eran del interior del país 80% (20 pacientes).
Se objetivó la presencia de 14 eventos trombóticos, de los cuales 10 fueron en territorio venoso, 1 en territorio arterial y 3 se presentaron con trombosis de pequeños vasos.
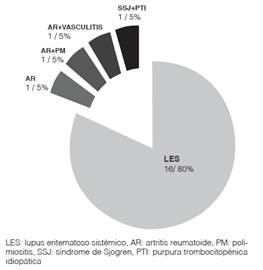
Se encontraron 20 pacientes con SAF y enfermedades autoinmunes asociadas, siendo la más prevalente el lupus eritematoso sistémico (16/20 pacientes (80%)). En la Figura 1 se observa la distribución de las enfermedades asociadas con el diagnóstico de SAF en la población estudiada.
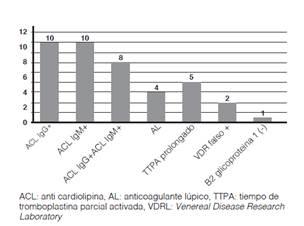
En relación a la determinación de los anticuerpos, se objetivó la siguiente distribución. aCL IgG(+):10 pacientes, aCL IgM(+):10 pacientes, 8 tenían ambos. AL: 4 pacientes, 3 pacientes c/ aCL(+).TTPA prolongado 5 pacientes:1c/ AL (+), 2 aislada y 2 c/ ACL(+) y AL(+). VDRL falso(+ ): 2 pac c/ ACL(+), AL(+) y TTPA prolongado presentes. B2 glicoproteína: se realizó en 1 paciente (-) (Figura 2).
Las manifestaciones clínicas objetivadas se presentan en la Tabla 1.
Tabla 1 Distribución de la población según las manifestaciones clínicas.
| Localización | nº casos |
|---|---|
| Sistema nervioso | |
| Accidente cerebro vascular isquémico | 8 |
| Neuromielitis óptica | 1 |
| Sx .Símil Esclerosis Múltiple | 1 |
| Cardiovascular | |
| Tromobosis venosa profunda | 7 |
| Infarto agudo del miocardio | 1 |
| Vegetación valvular | 1 |
| Isquemia de miembros inferiores | 1 |
| Hematológicas | |
| Trombocitopenia | 2 |
| Anemia Hemolítica | 5 |
| Cutáneas | |
| Livedo Reticularis | 1 |
| Ulcera en miembros inferiores | 1 |
| Hemorragia Subungueal | 1 |
| Isquemia digital | 1 |
| Pulmonares | |
| Sindrome de distres respiratorio | 1 |
Otros anticuerpos determinados fueron: ANA (+) en 21 pacientes, de los cuales 5 tenían AntiDNA+ y con trombocitopenia 2 pacientes y 5 con Test de Coombs directo (+), lo cual correspondió a una anemia hemolítica autoinmune (AHAI).
En relación a los fallecimientos, se registraron dos casos, un paciente de 17 y otra de 22 años por shock séptico y falla multiorgánica respectivamente.
En relación al tratamiento recibido se constató el uso de anticoagulantes orales (ACO) en 19 pacientes, ácido acetilsalicílico en 9 pacientes y heparina de bajo peso molecular en 1 paciente. Recibieron glucocorti coides 22 pacientes (prednisona 16 pacientes, bolos de metilprednisolona 6 pacientes). En relación al tratamiento inmunosupresor se constató el tratamiento con cloroquina/hidroxicloroquina en 16 pacientes, ciclofosfamida en 7 pacientes, azatioprina en 6 pacientes, metotrexate en 3 pacientes, leflunomida en 1 paciente y gamma globulina endovenosa (400mg/Kp/d,5 d.) en 1 paciente.
DISCUSIÓN
En el presente estudio de pacientes paraguayos con diagnóstico de SAF, se constató un clara predominancia del sexo femenino con el 92% (23/25) con una media de edad de 37,8 años, de forma coincidente con estudios previos3.
De forma interesante, se objetivó en la presente se rie de pacientes, que un 80% (20/25 pacientes) de los casos estaban asociados a una enfermedad autoinmune, por lo que requirieron además tratamiento inmunosupresor de acuerdo a la enfermedad de base. Este dato no está en concordancia con lo objetivado en el Euro-Phospholipid Project, donde se objetivó que un 53% de los pacientes presentaba un SAF primario. Posiblemente esta diferencia esté relacionada con el diferente número de pacientes analizados y tal vez con el origen europeo de los mismos. Nuevos estudios con un mayor número de pacientes deberán realizarse para poder corroborar estos datos3.
En relación a las manifestaciones clínicas, se observó que las manifestaciones más frecuentes fueron el accidente cerebro vascular isquémico y la trombosis venosa profunda. Estos datos están en concordancia con estudios previos donde se ha constatado que tanto la trombosis venosa profunda como el ACV isquémico han sido las manifestaciones más frecuentemente registrados3).
Esta revisión de casos de pacientes con SAF de origen paraguayo, ayudará a conocer las características de este síndrome en nuestra población, además de servir como base para futuros estudios.

