











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La púrpura de Schönlein Henoch fue descrita por primera vez en 1802 por Heberden, y reconocido como la asociación de púrpura y artralgias por Schönlein en 1837. En 1874, Henoch añadió la presencia de síntomas gastrointestinales, y la afectación renal en 18991. En 1968, Berger et al describieron 25 pacientes con hematuria recurrente y depósitos mesangiales de IgA que superaban los depósitos de IgG. Este hallazgo fue innovador, entonces paso a llamarse enfermedad de Berger y desde 2012 es conocida como vasculitis por IgA2.
La púrpura de Schönlein-Henoch es una vasculitis intervenida por inmunocomplejos de IgA que influyen en los pequeños vasos, generalmente intenso que afecta en el 90% a los niños2,3. Aunque se desconoce la causa de la enfermedad, está claro que el sistema IgA juega un papel central en la fisiopatología. Se sospechaba que bacterias, virus o agentes parasitarios desencadenaban la enfermedad en individuos genéticamente propensos1.
Suele manifestarse como una tétrada: púrpura palpable, artritis, dolor abdominal y enfermedad renal. La púrpura aparece en el 100% de los casos, se caracteriza por lesiones palpables de color púrpura rojizo de 2-10 mm, concentradas en las caderas y extremidades inferiores. La afectación articular (50-85%) suele manifestarse como oligoartritis de los miembros inferiores. Los síntomas gastrointestinales (50-70%) se identifican principalmente con dolor abdominal que empeora con la ingesta, puede ir acompañado de rectorragia o melena2. El trastorno renal (40-50%), puede presentarse con hematuria visible o proteinuria asintomática, síndrome nefrótico (<5% de los casos), lesión renal aguda (poco común) e insuficiencia renal crónica que se presenta hasta en 25% de los pacientes4.
El diagnóstico se basa principalmente en criterios clínicos respaldados por hallazgos histopatológicos5. Una biopsia de piel revela una vasculitis leucocitoclástica que afecta a los vasos sanguíneos pequeños. El hallazgo más típico es la infiltración de neutrófilos en pequeños vasos sanguíneos de la dermis. Los estudios de inmunofluorescencia determinan la presencia de depósitos de IgA y, en menor medida, depósitos de C3. La nefroscopía de rutina suele mostrar hiperplasia mesangial aislada y la glomerulonefritis semilunar solo se muestra en casos graves. El diagnóstico diferencial debe enfocarse en niños con púrpuras de causa hematológica y, en adultos, con vasculitis sistémicas principalmente vasculitis asociadas a ANCA, crioglobulinemias y panarteritis nodosa2.
En cuanto al tratamiento, el 94% de los niños y el 89% de los adultos no necesitan tratamiento pues se da resolución espontánea, por lo que el objetivo principal es tranquilizar a los pacientes. El tratamiento con corticoides es controvertido. Revisiones sistemáticas proponen que los corticosteroides disminuyen el tiempo del dolor abdominal, reducen el peligro de intususcepción, falla renal y repeticiones2.
A pesar de ser una vasculitis autolimitada, hay recidivas en solo un tercio de los casos. Es el principal factor pronóstico: a mayor edad peor pronóstico y mayor riesgo de nefritis, persistencia del síndrome nefrótico y extensión de los depósitos de IgA a las paredes de los capilares6.
CASO CLÍNICO
Paciente de sexo femenino de 50 años, ama de casa, consulta con antecedentes patológicos personales de aborto espontáneo, crisis convulsivas desde los 15 años en tratamiento esporádico que no especifica, último episodio hace 8 años, poliartralgias y dolor lumbar crónico de 3 años de evolución manejado con analgésicos orales.
Ingresa por cuadro clínico de 72 horas de evolución caracterizado por de la aparición de lesiones cutáneas de aspecto purpúrico localizadas en muslo y piernas (figura 1), acompañadas de dolor abdominal tipo cólico de localización difusa, de moderada intensidad, con hematoquecia. Estos síntomas se presentaron 24 horas posterior a laparotomía exploratoria realizada por cuadro de abdomen agudo en donde evidencian líquido inflamatorio y realizan apendicectomía.
En paraclínicos de ingreso se evidencia hemograma con plaquetas normales, PCR 19 mg/dL, creatinina 0,52 mg/dL, urea 7,30 mg/dL, orina con proteínas 500 mg/dL y hematíes 111 por campo. En sospecha de vasculitis con probable afectación cutáneo-renal. Los complementarios realizados evidenciaron proteinuria de 24 horas: 2,4 g. Valores de complemento, anticuerpos antinucleares, ANCA, anticoagulante lúpico, anticuerpo anticardiolipina IgM, serología VIH, VDRL, hepatitis B y C: todos negativos.
Se realizó biopsia de lesiones cutáneas donde se evidencia vasculitis neutrofílica necrotizante de pequeños vasos. La biopsia renal evidenció 16 glomérulos viables, con lesión difusa y global, aumento de la matriz y celularidad mesangial, moderada proliferación endocapilar y mínimo infiltrado linfocitario intersticial. En la inmunofluorescencia directa se observan extensos depósitos mesangiales IgA positivo de intensidad fuerte, C3 positivo, concluyendo como nefropatía IgA (figura 2).
Por la asociación clínica de afectación renal (proteinuria, hematuria), compromiso gastrointestinal y cutáneo junto con la confirmación de biopsia renal y cutánea, se concluye en el diagnóstico de púrpura de Schönlein Henoch. La paciente recibió 3 pulsos de metilprednisolona intravenosa de 500 mg/día, seguido de corticoides orales 60 mg/día, lisinopril 10 mg/día, omega 3 cada 8 horas, con mejoría clínica y bioquímica.
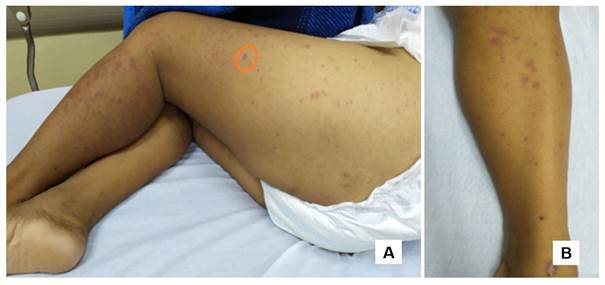
Figura 1 A. Presentación clásica de pápulas purpúricas en región de muslo izquierdo y pierna ipsilateral. B. Lesiones tempranas de pápulas rosadas que muestran pequeña necrosis central con formación de costras hemorrágicas.
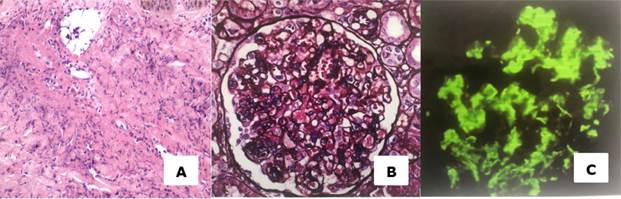
Figura 2 A. Histopatología de biopsia cutánea: se observan lesiones con evidencia de vasculitis neutrofílica necrotizante de pequeños vasos. B. Histopatología de biopsia renal: se observa aumento de la matriz y celularidad mesangial, moderada proliferación endocapilar y mínimo infiltrado linfocitario intersticial, presencia de depósitos mesangiales a nivel glomerular. C. En la inmunofluorescencia directa se detectan extensos depósitos mesangiales IgA positivo de intensidad fuerte, C3 positivo.
DISCUSIÓN
La púrpura de Henoch-Schönlein, conocida actualmente como vasculitis por IgA, se presenta en el 90% de los casos en niños de más de 10 años, con una incidencia de 3,0 a 26,7 de cada 100.000 jóvenes. Nuestro caso es llamativo ya que, en los adultos, la enfermedad sigue siendo poco común con una incidencia anual de 0,1 a 1,8 por cada 100.000 personas1,3,7,8.
Las numerosas observaciones clínicas han demostrado que la vasculitis por IgA puede seguir a una enfermedad infecciosa: se han descrito bacterias, virus y protozoos como posibles desencadenantes. En 2011, Suzuki et al. planteó la hipótesis de que la patogenia de la nefropatía por IgA se basa en cuatro aciertos: primero, la aparición de un proceso de glicosilación de IgA1 anormal que conduce a IgA1 deficiente en galactosa (Gd-IgA1); en segundo lugar, la formación de anticuerpos antiglicanos contra Gd-IgA1; tercero, la formación de inmunocomplejos circulantes nefrogénicos; y, cuarto, el depósito de estos complejos en el mesangio de los glomérulos que conduce a una lesión renal8-10.
En cuanto a la presentación inicial, el primer síntoma que manifestó la paciente fue la aparición de lesiones purpúricas. La púrpura cutánea es el aspecto más reconocido de la vasculitis por IgA. En un ensayo de 2016 se examinó a 260 adultos con vasculitis por IgA y al concluir determinaron que 100% de los pacientes presentaron púrpura. Estas lesiones comienzan como máculas eritematosas hasta llegar a púrpuras palpables en aproximadamente 10 días. El periodo de remisión de las lesiones es entre 10 y 14 días, y una resolución completa de las manifestaciones que va entre semanas a meses3,7,9,11. En nuestra paciente se presentaron lesiones papulares con necrosis central y formación de costras hemorrágicas. La literatura indica que las lesiones cutáneas de la vasculitis por IgA en adultos van a ser necróticas en 60% y esto conlleva a una mayor probabilidad de lesión renal11. La enfermedad renal se presenta en 62% de los casos, suele aparecer entre 1 a 90 días después de la erupción, pero puede aplazarse hasta seis meses3,7,9,12. Las manifestaciones digestivas en un adulto con vasculitis por IgA se dan en un 70% aproximadamente. El principal síntoma acompañante es dolor abdominal, presente en nuestra paciente que también tuvo hematoquecia. La hemorragia digestiva severa, la perforación intestinal y la invaginación son complicaciones potenciales2,9,13.
En 1990, el American College of Rheumatology estableció cuatro criterios para identificar la enfermedad: edad ≤20 años al inicio de la enfermedad, púrpura palpable, dolor abdominal y biopsia que muestra granulocitos en las paredes de pequeñas arteriolas o vénulas. Estos criterios tienen una sensibilidad de 87,1% y especificidad de 87,7% y pueden ser muy útiles en la población pediátrica, pero no son apropiadas en los adultos1. Nuestra paciente presentó 3 criterios de los 4 (solo no fue compatible la edad). La biopsia nos permitió un diagnóstico de certeza, por lo que es recomendable realizar la biopsia de piel dentro de las 24-48 horas iniciales de la presencia de púrpura, debido a que las reservas de IgA suelen desaparecer 48 horas después del comienzo. La biopsia renal está indicada en pacientes con proteinuria ≥500 mg/24h o creatinina >1,5 mg/dL. Nuestra paciente cumplía el criterio de la proteinuria. La biopsia, aparte de diagnóstica, guía en la decisión terapéutica. Los estudios demuestran que la presencia de hipercelularidad endocapilar está ligada a un peor pronóstico en ausencia de tratamiento inmunosupresor. La biopsia de piel muestra vasculitis leucocitoclástica y la biopsia renal depósitos de IgA en la inmunofluorescencia directa, con C3 positivo, tal cual se manifestó en nuestro caso3,13.
Los biomarcadores en la nefropatía por IgA han experimentado un gran impulso. El complemento puede estar disminuido (C3-C4), los valores de eritrosedimentación y proteína C reactiva no son útiles. El estudio de sedimento de orina permite demostrar el inicio de la afectación y es de utilidad para el seguimiento de la enfermedad. Entre los marcadores que podemos solicitar esta la proteinuria y a su vez los marcadores séricos creatinina y BUN para poder determinar la tasa de filtración glomerular. Se ha tratado de determinar marcadores biológicos para el diagnóstico y seguimiento, entre ellos tenemos: el sistema de complemento como depósito mesangial de la unión de lectina-manosa y C3, el sistema alterno de complemento con la participación de C3b, C3c, C5b9, la lectina unida a manosa o serina-proteasa 1. Pero hasta ahora, no existen marcadores bioquímicos que permitan concluir de forma fehaciente la existencia de la vasculitis por IgA. La biopsia renal sigue siendo el primer abordaje en la práctica clínica para el diagnóstico de la nefropatía por IgA3,7,10.
El diagnóstico diferencial se realiza con cualquier vasculitis que afecte a pequeños vasos. En 80% de estas se encuentran depósitos de IgA en la histopatología, así como también se puede observar IgA en: la hipersensibilidad por medicamentos, crioglobulinemia, lupus eritematoso sistémico, gammapatía monoclonal IgA8.
La vasculitis IgA se resuelve aproximadamente en 89% de los adultos. El tratamiento de la vasculitis depende de la histopatología y el objetivo que se quiera alcanzar. La erupción no requiere ningún tratamiento particular. Los fármacos mitigadores no esteroideos (AINES) y paracetamol son útiles para tratar las afectaciones articulares, pero deben evitarse en los casos de daño renal y gastrointestinal. El uso de corticoides está limitado en la vasculitis IgA con afectación renal. En la vasculitis IgA sistémica los datos de buena respuesta a la inmunosupresión suelen ser muy escasos, aunque se los suelen usar. Los efectos de la ciclofosfamida, azatioprina, micofenolato de mofetilo y ciclosporina A siguen siendo confusos3,7,14. Nuestra paciente recibió bolos de metilprednisolona por 3 días con buena respuesta terapéutica, yéndose al alta con dosis de prednisona 1 mg/kg/día. La literatura avala este tratamiento. Cabe recalcar que los pulsos de metilprednisolona (1 g/día durante 3 días) se guardan típicamente para los casos con asociación renal crítica. Sin embargo, la incertidumbre continúa. Algunos médicos consideran que un tratamiento de 6 meses con corticosteroides es el estándar de atención de rutina en cualquier paciente con vasculitis por IgA y proteinuria mayor de 1 g/24 h3,15. Hay varios criterios acerca del uso de corticoides, de momento no hay estudios clínicos que lleguen a un consejo para el uso de corticoides. En un ensayo controlado, aleatorizado, doble ciego, con placebo y corticosteroides realizado en 352 jóvenes con vasculitis por IgA de inicio tardío y con afectación renal negativa o menor halló que el tratamiento con corticosteroides no mostró ninguna ventaja sobre placebo en la disminución de la proteinuria un año después1,9. En otro estudio se compararon la combinación de micofenolato de mofetilo con prednisona en dosis baja y prednisona en dosis completa sola como terapia de inducción para la nefritis por IgA con proteinuria grande (>2 g/24h). Se incluyeron 53 adultos y se dividieron en dos grupos: pacientes que recibieron 1 g/día de micofenolato de mofetilo oral con dosis bajas de prednisona (0,4 a 0,5 mg/kg/día) y pacientes que recibieron dosis completas de prednisona (0,8 a 1 g/día). A los 6 meses, la tasa de remisión fue 76,9% en el grupo de prednisona de dosis completa y 55,5% en el grupo de micofenolato de mofetilo (diferencia no significativa)1,15.
Hernández Rodríguez et al. realizaron una revisión de literatura del uso de rituximab en adultos con vasculitis por IgA, acerca de las características de la enfermedad, la eficacia y la seguridad. Analizaron 20 estudios que incluían 35 pacientes con vasculitis por IgA tratados con rituximab. Casi el 90% de los pacientes tenían afectación renal antes del tratamiento con rituximab y la enfermedad resistente o refractaria a glucocorticoides u otros agentes inmunosupresores, principalmente con insuficiencia renal, fue el motivo de la administración de rituximab en 85,7% de los pacientes. El 94,3% de los pacientes presentó mejoría clínica de cualquier tipo y 74,3% logró una remisión sostenida al final del seguimiento. La administración de rituximab en pacientes con vasculitis por IgA se ha asociado con un buen perfil de seguridad5.
En cuanto al pronóstico, Van de Perre et al. demostró mediante un estudio retrospectivo en 85 pacientes adultos con vasculitis por IgA, con un seguimiento prolongado de 43 meses en 67 pacientes, que solo 49% logró una remisión completa. La afectación renal fue la manifestación orgánica persistente más común. El 43% tenía enfermedad recurrente y 27% experimentaron varias recaídas. A los 4 años de seguimiento, 21% experimentaron enfermedad progresiva y 34% enfermedad recidivante. El 7% desarrollaron nefritis después del diagnóstico, dentro de los primeros 6 meses de seguimiento. Al final del seguimiento, 15% tenían enfermedad renal crónica en estadio 3A 16.
No hay entendimiento claro de los mecanismos patogénicos desde el punto de vista celular o molecular, ni de las respuestas inmunitarias adaptativas o innatas que oriente al médico para poder obtener terapias dirigidas en estos casos. A pesar de que en los últimos años se han incrementado las investigaciones acerca de vasculitis por IgA, sigue siendo controversial el uso de corticoides y terapias inmunosupresoras. La justificación principal de utilizar corticoides es porque se está frente a una enfermedad inflamatoria20. A pesar de la evidencia limitada, los corticoides siguen siendo la piedra angular en el tratamiento con mejoría de la afectación renal como fue documentada en nuestro caso, y más si se usa pulsos de metilprednisolona con posterior uso de corticoides orales. El manejo no tan claro de la patología es de interés para que se instaure investigaciones terapéuticas que nos ayuden a evitar futuras complicaciones de la enfermedad.
Como conclusión, basada en la presentación clínica de la paciente, por sus manifestaciones de dolor abdominal, presencia de lesiones purpúricas más el hallazgo de alteración renal por pérdidas de proteínas en orina, al realizar un análisis exhaustivo del caso, lo primero que el internista debe intentar es llegar al diagnóstico de una vasculitis de manera específica para de esa manera aplicar un tratamiento juicioso y oportuno, para evitar daño de órganos vitales.

