











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
En los últimos 10 años en el Servicio de Oncología del Hospital Central del IPS fueron diagnosticados y tratados aproximadamente 8 casos de tumor carcinoide gástrico.
En Paraguay, en el periodo 2010 a 2014, el adenocarcinoma gástrico constituyó la cuarta causa de mortalidad por cáncer en hombres, que representa el 7.7%, mientras que en las mujeres es la séptima causa de mortalidad llegando a 4.7%, según datos disponibles en la página web de Dirección de Enfermedades no transmisibles de la Dirección General de Vigilancia de Salud (1, luego ya no se ha hecho ningún reporte. El tumor carcinoide corresponde a <1% de los tumores gástricos a nivel mundial, y lo mismo se refleja en Paraguay (2.
Se logró distinguir un tumor carcinoide de un adenocarcinoma en 1888, en el análisis descrito por Lubarsch; antes no se conseguía discernir con precisión la histología en informes previos realizados según reportes publicados (3.
Estos tumores son de crecimiento lento, pudiendo ser funcionantes y no funcionantes4. La manifestación clínica depende del tamaño tumoral, grado de invasión, localización, funcionalidad y tiempo de evolución (2. A veces el hallazgo del tumor ocurre durante una cirugía o los mismos síntomas pueden descubrirse durante la inducción anestésica (5.
Es característico en su forma sintomática el desarrollo del síndrome carcinoide conformada de diarrea, palpitaciones, sofoco, enrojecimiento facial, dolor abdominal intermitente y otros síntomas, hipotensión arterial y sibilancias (6. Se designa que la producción de serotonina está relacionada con los síntomas (2.
En ocasiones los síntomas se producen por el esfuerzo físico, por consumir productos ricos en tiramina como los quesos azules, vino o chocolate. Algunos medicamentos exacerban los síntomas (7,8.
El diagnóstico se efectúa por endoscopia digestiva, no presenta rasgos característicos, pero puede tener aspecto similar al de una úlcera, masa o pólipo (3.
Los exámenes que ayudan al diagnóstico son: búsqueda de niveles de serotonina en sangre, determinación de Niveles deA5HIAen la orina, TAC, RMN y el “octreoScan” (para identificar la mayoría de los carcinoides y otros tumores neuroendocrinos), siendo la inmunohistoquímica el patrón de oro para confirmar el diagnóstico con la positividad de los marcadores Cromogranina A y Sinaptofisina9.
El pronóstico varía si los tumores carcinoides cursan con o sin síndrome carcinoide, además la probabilidad de un segundo tumor primario aumenta en aquellos que presentan el síndrome carcinoide. La tasa de supervivencia disminuye si hay enfermedad metastásica10.
CASO CLÍNICO
Paciente masculino de 63 años con antecedente de enfermedad de Hansen, hipertensión arterial y diabetes mellitus tipo 2 en tratamiento; colecistectomizado 5 años antes por litiasis vesicular. En julio del 2019, consulta por disminución del apetito, saciedad precoz, sin náuseas ni vómitos de más de 1 año de evolución, con pérdida de peso (14 kg) en un mes y medio, dolor abdominal intermitente con cambios en el hábito defecatorio (más diarrea que constipación) y enrojecimiento facial.
Endoscopia digestiva alta (agosto 2018): mucosa del cuerpo gástrico proximal y fondo congestivo con lesiones elevadas entre 5 y 8 mm. Se toma biopsia que informa proliferación neoplásica, con escasa mitosis. Compatible con tumor neuroendocrino bien diferenciado.
Inmunohistoquímica (abril del 2019): tumor neuroendocrino Grado I. Cromogranina A, CD 56 y Sinaptofisina retornan positivas. El índice de proliferación ki 67 es menor al 2%.
Ecocardiografía (junio del 2019): FE:48 %. Hipertrofia ventricular izquierda, leve aquinesia latero-apical y antero-apical. Disfunción diastólica tipo I. VCI inspiratorio mayor al 50%.
En agosto de 2019 se realiza gastrectomía total con reconstrucción en Y de Roux, vaciamiento ganglionar D2. Anatomía patológica informa: NET bien diferenciado del cuerpo de estómago. Infiltra submucosa, menos de 2 mitosis por campo, no invasión vascular ni perineural. Márgenes libres. Ganglios 0/22.
TAC contrastada de cráneo, tórax, abdomen y pelvis (octubre del 2019): adenomegalias de hasta 11.3 mm en axila derecha y en la izquierda se visualizan linfonodos de hasta 10.4 mm. No evidencia de secundarismo encefálico, pulmonar, hepático ni óseo.
Diagnóstico definitivo: Tumor carcinoide gástrico Grado I; pT1, pN0, pM0, cM0, R0.
Tratamiento de mantenimiento: Octreotide 20 mg I.M cada 28 días desde hace 8 meses con buena tolerancia sin evidencia de progresión de la enfermedad.
En las figuras 1-4 se presentan las imágenes anatomopatológicas de mucosa gástrica y ganglio linfático.
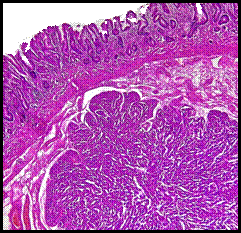
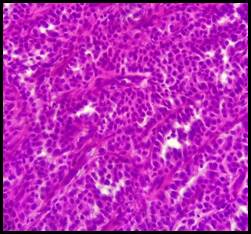
Figura 2 (HE 60x) Trabéculas de células neoplásicas uniformes, escaso citoplasma, núcleos redondos con cromatina dispersa.
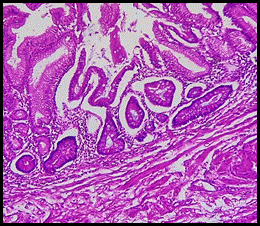
Figura 3 (HE 40x) Mucosa gástrica antral, focos de metaplasia intestinal con displasia de bajo grado.
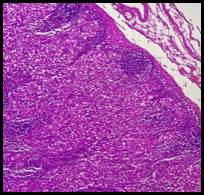
DISCUSIÓN
El tumor carcinoide gástrico, es una neoplasia rara y tiene un comportamiento distinto al adenocarcinoma gástrico; provienen de las células enterocromafínicas que se hallan en el fundus y cuerpo gástrico (8,11. Estas son las encargadas de producir histamina, estimulan la secreción de gastrina y del ácido clorhídrico en el estómago; en este tumor se describe una producción excesiva de la serotonina, sus metabolitos y otras sustancias como la somatostatina, provocando síntomas y signos conocidos como síndrome carcinoide, aunque también puede ser asintomático. Más raramente se ven estos síntomas en otros tumores de origen gástrico y son menos agresivos, como lo es en este caso, sin embargo, cabe resaltar que nuestro paciente presentó los síntomas típicos observados como en otros casos reportados (2.
Existen 3 tipos de tumores carcinoides gástricos:
El tipo I: es el más común, productora de hipergastrinemia, asociada frecuentemente a pólipos y gastritis atrófica crónica. Son casi siempre benignos. Puede plantearse una polipectomía endoscópica, en caso de cirugía gástrica debe realizarse una antrectomía para eliminar la hipergastrinemia y el estímulo del crecimiento celular12,13, lo cual se correlaciona con este caso mencionado, la opción quirúrgica fue considerada para cumplir con el propósito referido realizándose la gastrectomía total.
Se descartó tumor carcinoide tipo II y III por no cumplir con los criterios de ser portador del síndrome MEN 1. Tampoco presentó por endoscopía el hallazgo de úlceras con antecedente de resistencia al tratamiento convencional como ocurre en caso del síndrome de Zollinger Ellison 14,15) .
Según el grado, estos tumores bien diferenciados pueden ser de bajo grado, grado intermedio y alto grado (9 siendo los de bajo grado los más comunes, con índice mitótico <2/10 hpf (campo de alta potencia), índice de proliferación ki 67 < 3% similar a nuestro reporte.
En general, tanto en los tumores de bajo grado e intermedio el tratamiento estándar es la cirugía (15 y el uso de análogos de la somatostatina para controlar los síntomas16,17, mientras que en el tumor de alto grado o pobremente diferenciado la indicación terapéutica es la cirugía y quimioterapia17. Por ende, en este caso el tratamiento fue quirúrgico y octreotide para controlar los síntomas carcinoides. Además, la colecistectomía es recomendada cuando se pretende usar de forma prolongada el octreotide por riesgo frecuente de colelitiasis2, hacemos mención que el paciente ya había sido colecistectomizado 5 años antes por litiasis vesicular.
En muy bajo porcentaje se observa enfermedad carcinoide cardiaca es este tumor18, esto fue descartado en el paciente ya que sus alteraciones ecocardiográficas se relacionan a sus otras patologías de base.
CONCLUSIONES
Los tumores neuroendocrinos constituyen una entidad oncológica de difícil diagnóstico por la complejidad de los signos y síntomas que se enmascaran tras los tumores epiteliales gástricos, siendo la inmunohistoquímica el pilar principal para el diagnóstico.
Es importante identificar los síntomas que conforman el síndrome carcinoide maligno para tratarlo adecuadamente y evitar las complicaciones.
Para evitar un tratamiento agresivo se requiere individualizar la terapia según el tipo y grado histológico del tumor carcinoide.

