









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El Síndrome de Poliposis Juvenil (SPJ) es un trastorno autosómico dominante, raro y ocurre en 1 de 100.000 a 160.000 personas en la población general. Existe la forma familiar y la esporádica. Entre el 20% al 50% de los pacientes tienen antecedentes familiares de pólipos1,2,3,4.
Se manifiesta entre la primera o segunda década de la vida con múltiples pólipos juveniles. Los pólipos no se limitan al colon, sino que también pueden ocurrir en el intestino delgado y el estómago. Los criterios para establecer el diagnóstico de poliposis juvenil son:
1. Más de cinco pólipos juveniles en el colon o recto.
2. Cualquier número de pólipos juveniles en un paciente con antecedentes familiares de poliposis Juvenil.
3. Pólipos juveniles en cualquier parte del tracto gastrointestinal1,2,4.
En el SPJ, se han identificado mutaciones en los genes SMAD4 y BMPR1A, la presencia de defectos germinales en estos genes se halla en un 50 - 60 % de pacientes. Aproximadamente un 11-24% presenta mutaciones en SMAD4 y un 18-24% mutaciones en BMPR1A, y posiblemente un 5% en el gen PTEN. Este último probablemente tenga que ver con los síndromes de Cowden o de Bannayan Riley-Ruvalcaba (BRRS) y aún no hayan manifestado los síntomas. Una forma severa, a menudo fatal de SPJ denominada poliposis juvenil de la infancia se asocia con delecciones cromosómicas que afectan tanto a BMPR1A como a PTEN1-8.
PRESENTACIÓN DE CASOS
Caso 1: Mujer, de 14 años, con antecedente de sangrado rectal, color rojo rutilante, recurrente. Niega otros síntomas. Examen físico normal. En la rutina laboratorial se constata leve anemia. Tras la realización de colonoscopía se revela gran cantidad pólipos, distribuidos de forma difusa, de aspecto adenomatoso, algunos con erosiones, pediculados o subpediculados, desde el recto hasta colon sigmoides, no pudiendo progresar la colonoscopia más allá del sigmoides, debido a la obstrucción producida en la luz colonica. Mediante polipectomía endoscópica se extrajeron 2 pólipos (0,8 y 2,2 cm), que la anatomía patológica clasificó como pólipos juveniles.
Teniendo en cuenta los resultados endoscópicos e histológicos, fue intervenida quirúrgicamente, realizándose colectomía total videolaparoscópica. En la vista macroscópica de la colectomía total se observaron numerosas formaciones polipoides, pediculados y sésiles en el colon sigmoides y descendente (50 a 100), menor número (3 a 6) en el colon transverso, ascendente y ciego, que miden entre 0,2 a 2,4 cm. La histología mostró numerosos pólipos todos de tipo juvenil, hamartomatosos (Figura 1 y 2), y algunos presentaron displasia de bajo grado (Figura 3). El margen quirúrgico distal estaba comprometido por los pólipos.
La evolución de la paciente en el posoperatorio inmediato, fue excelente. A los 4 meses de la operación, sigue el control de los pólipos restantes en el recto.
Caso 2: Varón, de 12 años, que consultó por rectorragia tras la intervención quirúrgica de su hermana (caso anterior). Examen físico normal. Se realizó colonoscopía donde se observaron gran cantidad de pólipos, distribuidos de forma difusa, de aspecto adenomatoso, de tipo tubular alargado, algunos con erosiones en cabeza, pedículo y subpedículos, desde el recto inferior hasta el colon sigmoides, y más aisladas en colon descendente, transverso hasta el ángulo hepático. Mediante polipectomía endoscópica se extrajeron 2 pólipos (0,8 y 1,7 cm), que la anatomía patológica clasificó como pólipos juveniles.
Se realizó una endoscopía alta donde se encontraron varios pólipos de aspecto adenomatoso (entre 1,5 a 2 cm de diámetro) a nivel de la mucosa antral. Se realizó polipectomía y el diagnóstico anatomopatológico fue de pólipos juveniles.
Teniendo en cuenta los resultados endoscópicos e histológicos, fue intervenido quirúrgicamente, realizándose colectomía total. En el espécimen quirúrgico de la colectomía total se observaron numerosas (50 a 100) formaciones polipoides, pediculados y sésiles en el colon sigmoides, descendente, transverso y tres formaciones polipoides en ascendente, que miden entre 0,4 a 1,5 cm. La histología confirmó pólipos de tipo juvenil, hamartomatosos, sin displasia. El margen quirúrgico distal estaba libre.
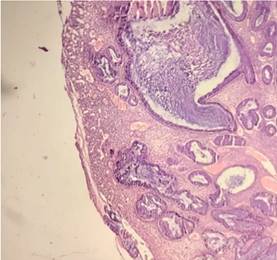
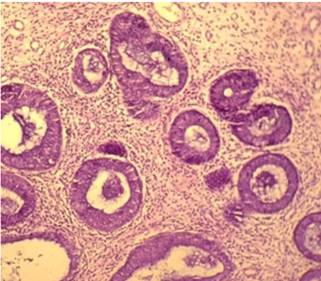
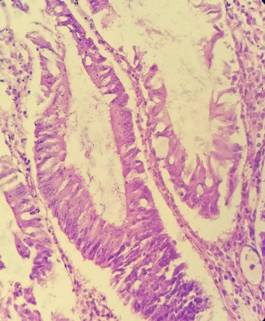
DISCUSIÓN Y CONCLUSION
Los pólipos juveniles son proliferaciones intestinales benignas, frecuentes en la infancia. Aparecen generalmente en el primer año de vida y raras veces persisten después de los 15 años. Aproximadamente el 30% se encuentra en el colon descendente, sigmoides y recto. Ocasionalmente se detectan en familias con múltiples pólipos en el colon, en las cuales no suelen presentar displasia, pero cuando las hay tienen mayor riesgo de desarrollar, adenomas y posteriormente cáncer colorectal (17 - 38%).1,2,4,5
Sachatello et al subdividieron el SPJ en tres grupos fenotípicos en relación a su presenta ción clínica: la poliposis juvenil de la infancia, la poliposis juvenil colónica (afectación colónica exclusiva), y la poliposis juvenil generalizada.
La poliposis juvenil de la infancia es la forma más grave de la enfermedad y con peor pro nóstico. La enfermedad, caracterizada por su presentación precoz en la infancia, se manifiesta en forma de diarrea sanguinolenta, enteropatía perdedora de proteínas, hipoproteinemia, ane mia, anasarca, intususcepción y prolapso rectal. Otras manifestaciones asociadas a este subtipo incluyen la macrocefalia, los dedos en palillo de tambor y la hipotonía. No existe historia fami liar.
La poliposis juvenil colónica y la poliposis juvenil generalizada habitualmente se manifies tan en la primera y segunda décadas de la vida en forma de sangrado rectal, prolapso de pólipo rectal, dolor abdominal, diarrea y anemia. Los estudios de seguimiento sugieren que los sín tomas o la anemia se manifiestan antes de la progresión a malignidad.
En relación a la edad de manifestación, en una serie de 218 pacientes, Coburn observó que la poliposis juve nil colónica se presentaba entre los 5-15 años de edad, (similar a nuestros casos presentados) mientras que la poliposis juvenil generali zada lo hacía a una edad más precoz. Hofting et al, en una revisión de 272 pacientes, observaron una afectación en orden de frecuencia del colon y recto (98%), estómago (14%), yeyuno-íleon (7%) y duodeno (2%).
Esta patología es muy grave, de peor pronóstico y produce más síntomas hemorrágicos (anemia e hipoproteinemias) en la infancia1,2,4,6,7.
Hasta ahora no existen marcadores clínicos, patológicos, inmunohistoquímicos o moleculares que permitan distinguir las dos formas de SPJ. El manejo de estos pacientes y sus familiares en riesgo se basa en la opinión de expertos debido a la ausencia de estudios caso-control. Es importante el estudio genético oportuno el grupos de riesgo. No existen estrategias de prevención dietético-farmacológicas5,6,7,8.
La vigilancia gastrointestinal alta debe efec tuarse preferencialmente mediante enterosco pia, aunque el uso de la cápsula endoscópica también puede ser de utilidad.
Los pacientes con un número pequeño de póli pos pueden ser tratados mediante polipectomía endoscópica. Si un pólipo con displasia de alto grado no puede resecarse completamente, o si se detecta un adenocarcinoma invasivo, debe efec tuarse una colectomía. En aquellos pacientes con un elevado número de pólipos no suscepti bles de tratamiento endoscópico debe efectuarse una colectomía total con anastomosis ileorrectal, o una proctocolectomía total con anastomosis ileoanal si el recto se halla muy afecto. En ambos casos debe efectuarse seguimiento del segmento colónico residual. En relación a los pólipos en el tracto alto, éstos deben ser tratados endoscópi camente. No obstante, en algunos casos es nece sario el abordaje quirúrgico9,10,11.