









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Los tumores oncocíticos de la glándula suprarrenal son raros, y muy pocos casos han sido descritos hasta el día de hoy, generalmente como casos aislados. Pocas publicaciones de estos tumores mostraban signos de virilización, el resto fueron no funcionantes.
Células similares a los oncocitos pueden verse en otros tumores córtico-suprarrenales no oncocíticos: en carcinoma de células renales que infiltran la suprarrenal, bien por invasión local o bien por metástasis, e incluso en feocromocitomas.
La diferenciación entre células corticosuprarrenalesoncocíticas y las células de estos tumores es difícil, y más si cabe, cuando estamos estudiando especímenes provenientes de punción aspiración donde el material obtenido es escaso.
CASO CLÍNICO
Mujer, 54 años, conocida asmática con tratamiento irregular, consumidora de corticoides diagnosticada de hipertensión arterial y diabetes mellitus 6 meses antes, en tratamiento con 3 antihipertensivos (valsartan/amlodipino/atenolol) y antidiabéticos orales (metformina 850 mg en 2 tomas).
Examen físico: Peso: 79 kg, Talla: 1.54 cm, IMC: 33.31, Presión Arterial: 190/100 mm Hg. Fascies rubicunda, redonda, hirsutismo de predominio en región facial, obesidad central, estrías violáceas en abdomen, hematomas en brazos. la misma refiere fatiga fácil y debilidad muscular.
Laboratorio: Glicemia: 108 mg/dL, HBA1c: 7.1%, TSH 0.88 uU/l (0.4 - 4), fT4 0.96 ng/dL (0.89 - 1.97), cortisol AM 27.7 ug/dL (6.2 - 19), ACTH inf a 1 pg/mL (7.2 - 63.3), Cortisol libre urinario 813 ug/24 hs (36-137), Cortisol libre urinario post dexametasona 779 ug/24 hs, hemograma, función renal y hepática en rango.
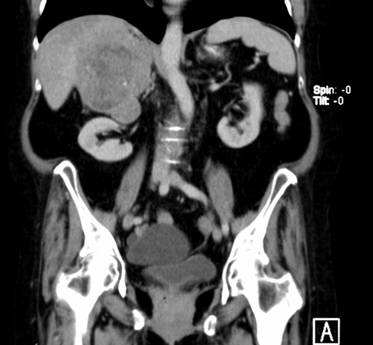
Se realiza una TAC de abdomen con contraste en el que se informa tumoración de aproximadamente 10x 12 cm en hígado. Se realiza una RMN de abdomen con contraste la cual informa voluminosa masa suprarrenal derecha, con síndrome de masa y compresión sobre el hígado, que no impresiona infiltrado, pero sin imagen de clivaje con el sexto y séptimo segmento, masa bien delimitada de diámetro AP 9.5, T de 8.6 y L 11 cm.
Demás valores laboratoriales(obtenidos tras 15 días de cambio de medicación antihipertensiva):Metanefrinas en orina 1.73 mg/24 hs (inf a 1 mg/24 hs), aldosteronemia 69 ng/dl (4 -31), DHEA-SO4 superior a 1500 ug/dL (29.7-182.2), Androstenediona superior a 10 ng/ml (0.3-3.3).
Se ajusta medicación antihipertensiva, por cifras tensionales de difícil control y se remite la paciente para evaluación para cirugía.
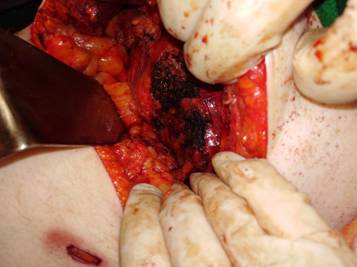
Se decide realizar una Suprarrenalectomíapor vía laparoscópica, constatándose durante la disección la infiltración de la misma a la vena cava inferior y sangrado tumoral en napa que no cede con maniobras hemostáticas por lo que se decide su conversión a convencional, se realiza la hemostasia correspondiente, se extrae muestras para biopsias, se deja drenaje aspirativo en el lecho y se da por concluido el acto quirúrgico. (Figura 1)
La paciente es trasladada a la unidad de cuidados intensivos, donde queda internada por 20 días, tiempo en el que obita como consecuencia de una sepsis a punto de partida pulmonar.
Anatomía patológica:
Se remiten para estudio histopatológico dos fragmentos irregulares de tejido de 40 x 25 x 15 mm y de 35 x 15 x 10 mm de diámetros, etiquetados como fragmentos de tumor suprarrenal derecho. Los mismos tienen una consistencia sólida, pero son blandos al ser levantados y friables. Su coloración es beigeanaranjada. Al corte los mismos muestran un tejido sólido, anaranjado homogéneo, apreciándose algunas áreas blanquecinas de pequeño tamaño. No se observan zonas quísticas o hemorrágicas (Figura 2).
El examen microscópico muestra una proliferación difusa de células de citoplasmas eosinófilos (oncocíticas) en el 100% de la tumoración no apreciándose células claras. Las células tienen bordes citoplasmáticos bien definidos y núcleos hipercromáticos, marcadamente atípicos, con anisocariosis, e imágenes de pseudo inclusión nuclear (grado III-IV/IV). No se observan figuras de mitosis. Se observan invasión capsular focal y extensos focos de necrosis coagulativa sobre los que se hallan sobre impuestos focos de licuefacción (Figura 3).
Diagnóstico anatomopatológico final: Carcinoma adrenocortical variante oncocítica.
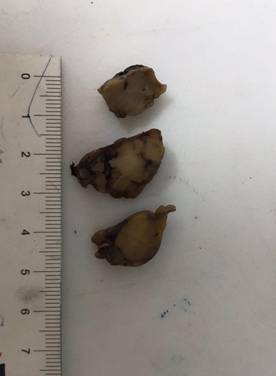
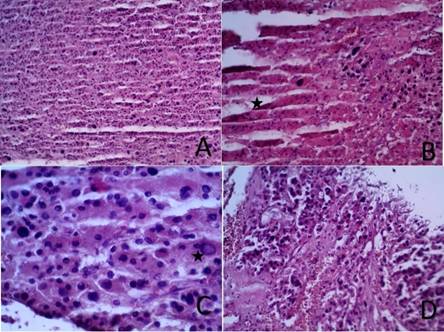
Figura 4. Microscopía. A. Proliferación difusa de células de citoplasmas eosinófilos (oncocíticas) en el 100% de la tumoración no apreciándose células claras (HE 10X). B. Extensos focos de necrosis coagulativa sobre los que se hallan sobre impuestos focos de licuefacción. En la imagen se muestra la transición (zona de necrosis marcada con una estrella) (HE 20X). C. Las células tienen bordes citoplasmáticos bien definidos y núcleos hipercromáticos, marcadamente atípicos, con anisocariosis, e imágenes de pseudo inclusión nuclear (grado III-IV/IV) la que se muestra con la estrella (HE 40X). D. Se observa invasión capsular focal (HE 20X).
DISCUSIÓN
Los tumores suprarrenales son muy comunes y afectan entre el 3% y el 10% de la población humana, y la mayoría son adenomas corticosuprarrenales (AAC) benignos y no funcionales. El carcinoma (ACC) por el contrario, es una enfermedad muy rara, con una incidencia de aproximadamente uno o dos casos por millón de habitantes por año, representando del 0.05% al 0.2% de todas las malignidades.1
Hay una distribución de edad bimodal, con un pequeño pico en las dos primeras décadas y un pico más grande en la quinta década.2
Se han notificado carcinomas adrenocorticales en aproximadamente el 1% de los pacientes con el síndrome de Li-Fraumeni, y la mayoría de los individuos afectados presentan mutaciones de p53 en el locus cromosómico 17p13. De hecho, estos tumores pueden ser la única manifestación de este trastorno en la infancia. La frecuencia de tumores malignos corticales también aumenta en pacientes con síndrome de Beckwith-Wiedemann (BWS) e hiperplasia suprarrenal congénita.2,3
Los carcinomas adrenocorticales se desarrollan algo más comúnmente en mujeres que en hombres en la mayoría de las series clínicas grandes, aunque algunos estudios han demostrado un ligero predominio masculino.
Algunos pacientes pueden presentar dolor abdominal, y hasta un 30% puede tener una masa abdominal palpable.2,4
Estos tumores pueden estar asociados con el síndrome de Cushing o evidenciar sobreproducción de esteroides sexuales, y los síndromes mixtos ocurren con mayor frecuencia que con los adenomas corticales. En raras ocasiones, la producción de mineralocorticoides puede estar presente. Una proporción significativa de carcinomas corticales (hasta 75% en algunas series) puede no asociarse con síndromes de sobreproducción de hormonas. En algunos casos, los pacientes pueden mostrar signos de hipoglucemia como resultado de la producción de factores de crecimiento similares a la insulina por el tumor o hipercalcemia como resultado de la producción del péptido relacionado con la hormona paratiroidea.3
Como regla general, los carcinomas corticales son tumores grandes que pesan más de 100 g en adultos; lo más frecuente es que el peso tumoral supere los 750 g. En raras ocasiones, sin embargo, los tumores que pesan menos de 50 g metastatizan, mientras que una pequeña proporción de tumores que pesan más de 1000 g pueden no hacerlo. Debido a este hecho se ha dejado de utilizar el peso como predictor de malignidad.
En el momento del diagnóstico, la evaluación inicial debe incluir un examen físico minucioso e historial del paciente con particular énfasis en los síntomas y signos de producción hormonal excesiva. Los pacientes deben someterse a una evaluación bioquímica básica que incluya creatinina, pruebas de función hepática y un hemograma completo. Estos valores guiarán la terapia adicional y el manejo de la enfermedad. Una evaluación hormonal inicial es crucial. La estadificación debe incluir como mínimo una tomografía computarizada (TC) o una resonancia magnética (RM) del abdomen, pelvis y una TC del tórax. Otras imágenes deben guiarse por la sospecha clínica (por ejemplogammagrafía ósea en busca de metástasis esqueléticas). Un enfoque en la historia familiar es esencial para identificar posibles contribuciones hereditarias.4,5
El abordaje quirúrgico de esta patología puede ser realizado tanto por via convencional como laparoscópica, obteniéndose similares resultados en cuanto a resecabilidad oncológica.6
En cuanto a la anatomía patológica de los tumores adrenocorticales ha avanzado sustancialmente en las últimas 4 décadas. Inicialmente, se pensó que el tamaño del tumor era el principal factor para determinar qué tumores poseían potencial maligno y, en consecuencia, se podían clasificar como ACC. Aunque el tamaño tumoral aún posee importancia diagnóstica, los algoritmos diagnósticos actuales han evolucionado para incorporar una variedad de parámetros clínicos, histológicos e inmunohistoquímicos. Para distinguir entre benignidad y malignidad en los tumores córticosuprarrenales. Weiss y cols. plantearon un sistema que incluía 9 criterios histológicos: intensa atipia nuclear, citoplasma eosinófilo (>75%), arquitectura difusa (>33%), presencia de necrosis, mitosis (>5 por 50 campos de gran aumento), mitosis atípicas, invasión capsular, venosa y sinusoidal. La presencia de 3 o más criterios sugieren que se trata de un carcinoma. Sin embargo, en los tumores córticosuprarrenalesoncocíticos este sistema no es aplicable, ya que por definición muestran ya tres de estos criterios, por lo que se debe ser muy cuidadoso en el diagnóstico de carcinoma. De cualquier manera, si bien nuestro caso presenta por definición los tres criterios de los tumores oncocíticos (intensa atipia nuclear, citoplasma eosinófilo en el 100% de las células y arquitectura difusa en el 100% del tumor), presenta además otros criterios enumerados por Weiss que son la presencia de necrosis extensa, la invasión capsular y la invasión venosa.3,7,8
El diagnóstico diferencial por métodos inmunohistoquímicos se muestra en la tabla 1 y los criterios de malignidad en la Tabla 2.8
La inmunohistoquimica en este caso reportó un tumor oncocítico suprarrenal Calretinina(, MelanA (, Inhibina(, AE1/AE3 (/-.
En conclusión, presentamos un caso de un carcinoma suprarrenal oncocíticofuncionante, tumor de muy baja incidencia, que plantea problemas en su diagnóstico y pronóstico, si bien existen una serie de criterios histológicos que nos ayudan a la interpretación. En este caso el tumor ha sido irresecable, debido a la infiltración en estructuras adyacentes incluida la vena cava.
Tabla 1: Diagnóstico diferencial inmunohistoquímico de los tumores suprarrenales.
| Feocromocitoma | SF+, Cra+, CD56+, Vimneg (+ en 25% de casos), S100+, NF+ |
| Adenoma o carcinoma adrenocorticaloncocítico | Vim+, EMA neg, CEA neg, Inhibina+, Melan A+, SF+, Craneg, Calrretinina+ focal |
| Oncocitoma renal | Cam 5.2+, CK7 neg, CK20 neg, Vim neg, CD10+/- |