










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkCirugía paraguaya
On-line version ISSN 2307-0420
Rev. Cir. Parag. vol.37 no.1 Asunción June 2013
Reporte de caso
Feocromocitoma asintomático
Asymptomatic pheochromocytoma
Ramírez Sotomayor, Julio 1; Pfingst, Carlos2 ; Gutirrez,Tomás2; Vera, Hugo2
RESUMEN
El feocromocitoma es un tumor que produce catecolaminas, mayor responsable de la hipertensión arterial en jóvenes, con mayor frecuencia se localiza en la medula suprarrenal, unilateral. Generalmente asociado con neoplasia endócrina múltiple. Los pacientes presentan una serie de síntomas, entre ellos la hipertensión arterial.
Se presenta el caso de una paciente de 31 años, con dolor en hipocondrio derecho de un año de evolución, tipo pesadez y pérdida de peso de 3 kg. Al examen físico se constató tumoración en hipocondrio derecho que llega a epigastrio, de 12 cm de diámetro. Con el diagnóstico de tumor retroperitoneal obtenido por imágenes se decide una laparotomía exploradora donde se visualizó un tumor de 18cm. de diámetro, sólido, en polo superior del riñón derecho, adherido a vena cava. Se realizó una exéresis tumoral y el informe de anatomía patológica confirmó feocromocitoma.
Palabras Claves: Catecolaminas, Médula suprarrenal, Neoplasia endócrina múltiple.
SUMMARY
Pheochromocytoma is a catecholamine-producing tumor, largely responsible for hypertension in young, most often located in the adrenal medulla, unilateral. Generally associated with multiple endocrine neoplasia. Patients present an array of symptoms, including hypertension.
A case of a 31 years with right upper quadrant pain a year of evolution, such heaviness and weight loss 3 kg. Physical examination found a mass in the right upper quadrant reaching epigastrium, 12 cm in diameter. With the diagnosis of retroperitoneal tumor images obtained by exploratory laparotomy decides where a tumor was visualized 18cm. in diameter, solid, in the upper pole of the right kidney, vena cava joined. Tumor resection was performed and the pathology report confirmed pheochromocytoma.
Keywords: Catecholamines, Adrenal medulla, Multiple endocrine neoplasia.
INTRODUCCIÓN (1-5)
El feocromocitoma fue descrito por Frankell en 1886 en las suprarrenales de un niño que murió de shock. El feocromocitoma es un tumor del sistema nervioso simpático que se desarrolla a partir de las células cromafines y producen catecolaminas (noradrenalina y adrenalina) de manera excesiva. Es un tumor que tiene una frecuencia de 1 a 2 por cada 100000 habitantes por año sin diferencias entre sexos y su pico de máxima incidencia es entre la tercera y cuarta década de la vida. Es responsable del 0,1 al 1% de las hipertensiones arteriales. Aproximadamente el 90% de los casos se localizan en la médula suprarrenal y el 10% restante es de ubicación extra suprarrenal, mayoritariamente intraabdominales, en particular en el órgano de Zuckerkandl. En el 80 - 90% de los casos es unilateral, con predominio en el lado derecho, sin conocerse la causa. El 10% de los casos es maligno, siendo la presencia de metástasis el único criterio de malignidad, dado que no hay características histopatológicas que la definan.
Los pacientes con feocromocitoma presentan una serie de síntomas que van desde una hipertensión lábil moderada, hasta la muerte súbita por crisis hipertensiva, infarto de miocardio o accidente vascular cerebral. Los síntomas clásicos son episodios paroxísticos de cefalea intensa, hipertensión arterial, taquicardia y sudoración profusas. En el 50% de los casos la hipertensión es sostenida, mientras que es intermitente en el resto. Por regla general, existe pérdida de peso, pero la obesidad no excluye el feocromocitoma. Son frecuentes los síntomas y signos Son frecuentes los síntomas como aumento de sudoración. La hipotensión ortostática es una consecuencia de la reducción de volumen plasmático debido al exceso de estimulación adrenérgica.
La ecografía es un buen método para detectar el tumor por que suelen tener un buen tamaño, ser redondeados, hipoecoicos y homogéneos, excepto en casos de hemorragia o necrosis intratumoral.
CASO CLÍNICO
Paciente de sexo femenino de 31 años de edad que acudió por dolor en hipocondrio derecho de un año de evolución al inicio era leve y 8 días antes del ingreso el dolor gana en intensidad, tipo pesadez, y no cede a la ingesta de analgésicos, además de pérdida de peso de 3 kilogramos aproximadamente. Niega nauseas y vómitos. Niega ictericia, coluria y acolia y sensación febril.
Al examen físico se constató tumoración en hipocondrio derecho que llega a epigastrio de borde bien definidos, lisa, de 12 cm de diámetro aproximadamente, no doloroso, no pulsátil, que excursionaba con los movimientos respiratorios. El resto de órganos y sistemas sin particularidades.
Los estudios laboratoriales realizados fueron normales.
En la ecografía se constató el riñón derecho aumentado de tamaño, con una imagen nodular mixta predominantemente sólida en polo superior del riñón derecho.
La tomografía abdominal observó una tumoración en retroperitoneo, tumoración heterogénea que abarca el hipocondrio y flanco derecho, en contacto con el hígado y el riñón (Fig. 1).
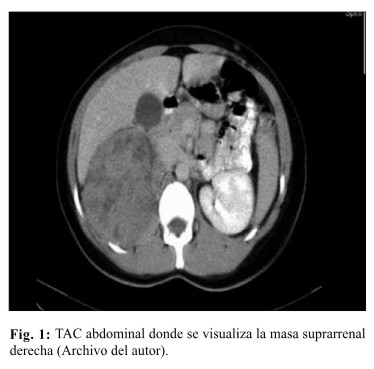
Con el diagnóstico de Tumor retroperitoneal de etiología a determinar, se decide una laparotomía exploradora a través de una incisión subcostal bilateral. Se visualizó un tumor de 18cm. de diámetro, sólido, en polo superior del riñón derecho, adherido a vena cava, (Fig. 2) donde se realizó una exéresis tumoral más drenaje de la cavidad abdominal.
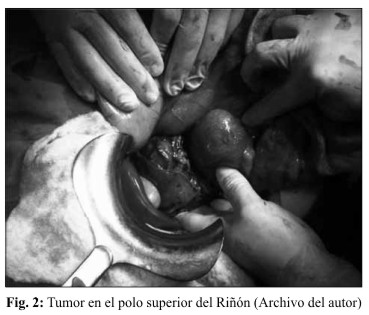
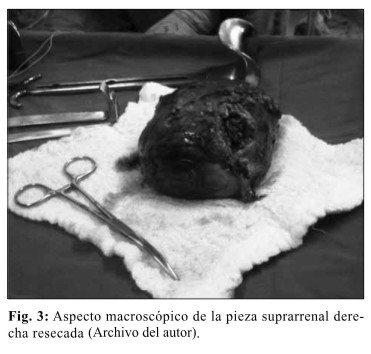
El informe de anatomía patológica: reveló feocromocitoma (paraglioma) de 10 cm del retroperitoneo, en la región de la glándula suprarrenal derecha resecado en su totalidad. (Fig. 3)
DISCUSIÓN
El feocromocitoma es un tumor raro de la médula suprarrenal, el cual puede segregar catecolaminas en exceso (1). Los signos y los síntomas del feocromocitoma pueden incluir hipertensión arterial, dolor (cefalea o dolor en los flancos, abdominal o torácico), hiperhidrosis, ansiedad o ataques de pánico, arritmias cardíacas o, muerte súbita. También puede ser detectado por la presencia de una masa incidental asintomática en un estudio por imágenes abdominal. (2)
Feocromocitomas familiares pueden aparecer en personas con síndromes genéticos: Von Hippel-Lindau (VHL) (3) ,Neoplasia Endocrina Múltiple (MEN – especialmente del tipo 1),Neurofibromatosis Tipo 1 (NF1), por lo tanto debe ser investigado en todas estas patologías. El estudio bioquímico de primera elección es la medición de normetanefrina y metanefrina libres en el plasma para guiar hacia el diagnóstico.(4)
En la mayoría de los casos el principal motivo de consulta es una hipertensión arterial en jóvenes que no presentan buena respuesta a los fármacos anti hipertensivos, por ellos se explora una causa secundaria utilizando los medios auxiliares de imágenes sospechando en un tumor supra renal (5). La TAC confirma o descarta la sospecha tumoral. Una vez objetivado el tumor se determina el grado de resecabilidad.
Los abordajes quirúrgicos parecen ser dos: la adrenalectomía laparoscópica y la cirugía abierta, dependiendo de la localización del feocromocitoma. La hipertensión arterial asociada es curable con la resección quirúrgica del tumor. Hay riesgo de muerte súbita debido a la descarga adrenérgica.
Por lo menos un 10% de los tumores son malignos. La detección en los casos de afección familiar puede resultar en el diagnóstico precoz de otros miembros de la familia. Para todos los casos la cirugía está indicada.
BIBLIOGRAFÍA
1. Recasens M, Oriola and col. Asymptonic bilateral adrenal pheochromocytoma in a patient with a germline v804M mutation in the RET proto-oncogene. Clin Endocrinol 2007; 67: 29-33. [ Links ]
2. Lavin N. Endocrinología y metabolismo. Madrid ed. Marban Libros, S.L; 2003.p.149-53 [ Links ]
3. Lenders J.W. and col., Pheochromocytomas in von Hippel- Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 2001; 86: 1999-2008 [ Links ]
4. Dluhy R, Lawrence J, Williams RH. Hipertensión de origen endocrino., Tratado de Endocrinología Clínica. RH Williams. 10ma ed. USA: Ed. Elservier; 2000; 677 [ Links ]
5. Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001 May;86(5):1999-2008. [ Links ]
1. Jefe de sala
2. Residente
Servicio de Cirugía General Hospital Nacional de Itauguá (Itauguá Guazú - Paraguay). Tel/ Fax: 0294/ 321450
Autor correspondiente: Dr. Carlos Pfingst - Intendentes Militares 939 dpto 13 C - Email: carlospfingst@gmail.com
Fecha de recepción: 28-julio-2012 Fecha de aceptación: 06-mayo-2013

