









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkIntroducción
El volumen de emisión a la atmósfera de los llamados compuestos orgánicos volátiles (COVs) es grande tanto de aquellos que provienen de fuentes biogénicas como de las antropógenicas (Atkinson & Arey, 2003), estos pueden reaccionar luego con diferentes elementos o compuestos y generar entidades moleculares que son potencialmente peligrosas para la salud de los seres humanos, por ello el estudio tanto teórico como experimental del comportamiento en la atmósfera de los mismos es incesante y numerosos trabajos han sido publicados en ese sentido en los últimos años (Atkinson & Arey, 2003); (Jiang et al., 2009); (Jiang et al., 2010); (Sandhiya et al., 2012); (Kunaseth et al., 2017); (Aazaad & Lakshmipathi, 2017); (Shaw et al., 2018).
Entre los COVs de interés se encuentran los alcoholes insaturados entre los cuales el 1-penten-3-ol es objeto de estudio del presente trabajo. Compuestos similares al mismo y el propio 1-penten-3-ol han sido estudiados en los últimos años (Papagni et al., 2001); (Orlando et al., 2001); (Jiménez et al., 2009); (O’Dwyer et al., 2010); (M. E. Davis & Burkholder, 2011); (Bernard et al., 2012) analizando las reacciones de éstos con el ozono, con el radical OH o con los nitratos, sin embargo muy pocos estudios de reacciones entre el 1-penten-3-ol y el cloro fueron llevados a cabo, destacándose entre ellos el llevado a cabo por Rodríguez y sus colaboradores (2010). En el presente trabajo se realiza un estudio de- tallado de la molécula del 1-penten-3-ol y de sus conformeros de rotación, así mismo se lleva a cabo un estudio de sus reacciones con átomos de cloro, se analiza la reacción de abstracción de uno de sus hidrógenos para la formación del hidruro de cloro y la adición del átomo de cloro al doble enlace del carbono en la misma, estos estudios se llevan a cabo utilizando los métodos de la Teoría del Funcional de la Densidad a un nivel de cálculo B3LYP/aug- cc-pVXZ (X=2, 3, 4).
Materiales y métodos
Fueron realizados cálculos para la optimización de la geometría de las diversas especies estudiadas y los estados de transición asociados con ellas, para la determinación de energías, y para el cálculo de frecuencias armónicas; en todos los casos fue empleada la Teoría del Funcional de la Densidad con el funcional B3LYP (Yang et al., 1988); (Becke, 1993) utilizando las bases aumentadas, polariza- das y de correlación consistente aug-cc-pVDZ y aug-cc-pVTZ; también fueron realizados cálculos puntuales de energía con el funcional mencionado y una base aug-cc-pVQZ (Dunning, 1989); (Kendall et al., 1992) (Woon & Dunning, 1993). En algunos casos los cálculos exploratorios fueron realizados a nivel DFT/B3LYP/321G+** (Pietro et al., 1982). En todos los casos fue utilizado el programa computacional Gaussian 03 (Frisch et al., 2004).
Para los cálculos de energía a base infinita (CBS) fueron utilizados varios modelos, ellos son el exponencial (Feller, 1993) (Halkier et al., 1999) dado por: 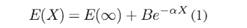 el modelo potencial (Nyden & Petersson, 1981); (Petersson et al., 1985) dado por:
el modelo potencial (Nyden & Petersson, 1981); (Petersson et al., 1985) dado por:  el modelo exponencial expandido (Peterson et al., 1993) dado por:
el modelo exponencial expandido (Peterson et al., 1993) dado por:  y los modelos de extrapolación de dos y tres parámetros (Martin, 1996) dados por:
y los modelos de extrapolación de dos y tres parámetros (Martin, 1996) dados por: 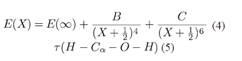
Para realizar la extrapolación a base infinita se usaron los valores de energía obtenidos por optimización de geometría a los niveles de teoría DFT/ B3LYP/aug-cc-pVXZ con X = D y T y cálculo de energía puntual con X = Q usando la geometría optimizada previamente con X = T.
Para la búsqueda de los conformeros de rotación se partió de la estructura mostrada en la Figura 1 y el ángulo diedro dado por 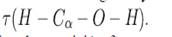 .
.
Los sistemas en estados de transición fueron optimizados a nivel DFT/B3LYP/aug-cc-pVXZ con X = D y T, utilizando indistintamente las herramientas TSBERRY y TS (QST2) y los mismos fueron verificados mediante el cálculo de las frecuencias armónicas de vibración y la verificación de la existencia de una sola frecuencia imaginaria. En todos los casos la conexión entre los reactivos y productos, con sus respectivos estados de transición, fueron confirmados siguiendo el correspondiente camino de reacción (IRC).
Resultados y discusión
Estudio de rotámeros
La Figura 2 muestra los resultados de la búsqueda de conformeros de rotación del 1-penten-3-ol, los cálculos para la misma fueron llevados a cabo con un nivel de teoría DFT/aug-cc-pVDZ y en cada caso se estudiaron 72 estructuras diferentes, rotando el ángulo diedro apropiado (mostrado en la Figura 1) cada 5°.
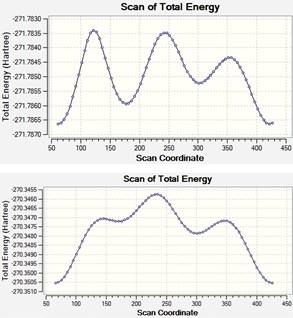
Figura 2 Búsqueda de conformeros de rotación del 1-penten- 3-ol, en el eje horizontal se muestran los valores del ángulo diedro rotado en grados sexagesimales.
Puede verse que en cada caso fueron halladas 3 estructuras con mínimos locales de energía, los ángulos diedros de estas estructuras son reportados en la Tabla 1.
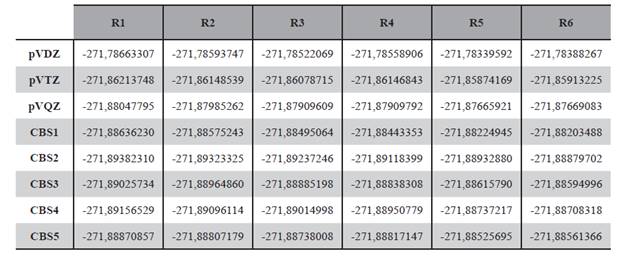
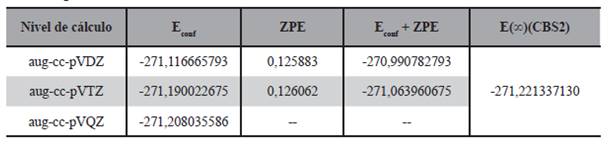
La geometría de estas seis estructuras fueron optimizadas a nivel DFT/B3LYP/aug-cc-pVDZ y DFT/B3LYP/aug-cc-pVTZ, también se realizó el cálculo puntual de energía de los mismos a nivel DFT/B3LYP/aug-cc-pVQZ; por otro lado se llevó a cabo el cálculo de energía con expansión a base infinita con los modelos mostrados en las ecuaciones (1), (2), (3), (4) y (5), los resultados de estos cálculos son presentados en las Tabla 2.
Se puede ver en todos los niveles y modelos de cálculo que el rotámero R1 es el de menor energía, seguido del rotámero R2. Las dos estructuras pueden apreciarse en la Figura 3.
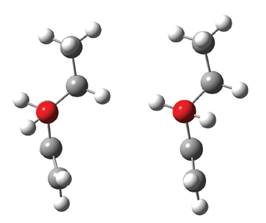
Estas dos estructuras fueron reportadas anteriormente por Rodríguez y sus colaboradores (2010), en este reporte al igual que en el presente trabajo el rotámero R1 es menos energético que el rotámero R2, sin embargo en dicho trabajo la diferencia de energía reportada entre ambos conformeros de rotación es de 1,9 kcal/mol con cálculos ab initio a nivel MP2/6-311G** mientras que en el presente trabajo para los distintos niveles de cálculo realizados la diferencia energética varía entre 0,38 kcal/ mol a 0,44 kcal/mol.
Por otro lado si comparamos las energías de los rotámeros, como podemos ver en la Figura 4, la energía de los mismos van disminuyendo en la medida que aumenta el tamaño de la base, también es posible notar que en la medida que estas bases aumentan de tamaño la diferencia entre las energías disminuye, estos resultados coinciden con los hallados recientemente por Ventura y su grupo de trabajo (Ventura et al., 2017).

Por otro lado y como puede verse en la Figura 5 de los modelos de cálculo de energía con proyección a base infinita, en todos los casos la energía calculada con el modelo dado por la ecuación 2 es la que resulta ser de menor valor.
También se puede apreciar comparando los mostrado en la Figura 4 y en la Figura 5 que la energía calculada con las diversas bases se aproxima al valor de la energía a base infinita en la medida que el tamaño de la base aumenta, en ese sentido se puede mencionar que para el rotámero R1 el valor de la energía con base triple Z es tan solo 0,012% mayor a la energía proyectada por el modelo CBS dado por la ecuación 2.
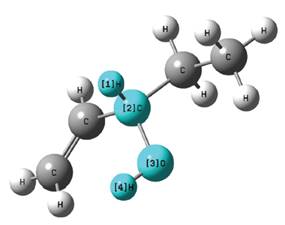
Teniendo en cuenta que el rotámero de menor energía hallado fue el denominado R1, con el mismo se realizó el resto del estudio. En ese sentido la geometría de dicha molécula fue optimizada a los niveles DFT/B3LYP/aug-cc-pVDZ y DFT/B3LYP/ aug-cc-pVTZ y las distancias interatómicas obtenidas se pueden apreciar en la Tabla 3 y la referencia de los mismos en la Figura 6.
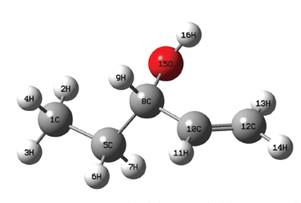
Figura 6 Imagen generada de la geometría del 1-penten-3-ol optimizada al nivel DFT/B3LYP/aug-cc-pVTZ.
Para la comparación con datos experimentales (Tabla 3) fueron utilizados los reportados por Lide (2004) los cuales fueron medidos en compuestos orgánicos similares al 1-penten-3-ol en fase gaseosa mediante una amplia gama de técnicas, los resultados del cálculo que se obtuvo en el presente trabajo y los valores de referencia experimentales tienen relativamente buena concordancia, los obtenidos de la optimización realizada al nivel doble Z, la mayor diferencia está en la longitud de enlace R(5,8) donde la misma es del 1,49 %, mientras que la mínima diferencia se encuentra en la longitud de enlace R(8,15) con un 0,08%.
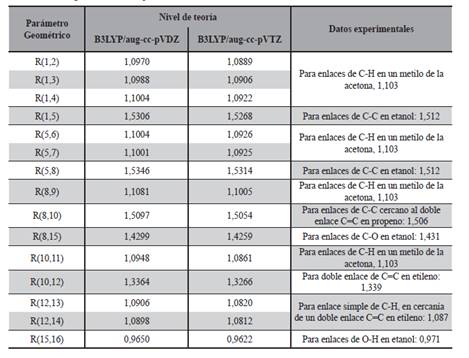
En cuanto a los resultados de la optimización a triple Z la máxima diferencia entre valores calculados y experimentales se halla en la distancia interatómica R (5,8), como en el caso anterior, con una diferencia de 1,28% y la mínima en la longitud de enlace R(8,10) con una diferencia de tan solo 0,04%. Al comparar las longitudes de enlace obtenidas (Tabla 3) en los dos niveles de cálculo utilizados, es decir a DFT/B3LYP/aug-cc-pVXZ, con X = D y T, se puede apreciar que estas distancias son todas menores en el nivel triple Z lo cual indica que el incremento en la captura de la energía de correlación electrónica, al aumentar el tamaño de la base, disminuye la distancia interatómica.
Por otro lado al comparar con el estudio teórico realizado por Rodríguez y colaboradores (2010) se puede notar que los resultados son muy cercanos, siendo el calculado en el trabajo mencionado la longitud del doble enlace R(10,12) de 1,340 Å mientras que nuestros cálculos arrojan 1,336 Å para el nivel doble Z y 1,327 Å para el nivel triple Z con una diferencia del 0,97 %, viéndose además que el nivel DFT/B3LYP/aug-cc-pVDZ se acerca más al nivel de cálculo utilizado en el trabajo mencionado (Tabla 3).
En cuanto al cálculo de las frecuencias armó- nicas del 1-penten-3-ol es interesante mencionar que Davis y Burkholder al medir la sección eficaz del 1-penten-3-ol reportan un pico de absorción en 2971,8 cm-1, un número de onda muy cercano al cálculo realizado en este trabajo que tiene un pico a 2937,2 cm -1, dando una diferencia de tan solo 1,2 % del valor experimental, frecuencia que de acuerdo con nuestra simulación correspondería a un modo de vibración relacionado con la variación de distancia entre átomos (M. Davis & Burkholder, 2011). En ese mismo sentido la literatura disponible reporta valores experimentales típicos de números de ondas para el estiramiento de los dobles enlaces de compuestos orgánicos, como el 1-hexeno, en 1660 cm-1, siendo el valor calculado en el presente trabajo de 1700 cm-1 una diferencia de tan solo 2,4 % respecto a los valores referenciados (McMurry, 2008; Stuart, 2004; Wade, 2011).
Reacción de abstracción
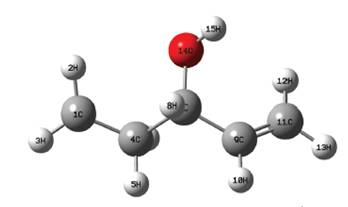
De todas las abstracciones posibles de los hidrógenos del 1-penten-3-ol fue completado el estudio a la que corresponde al hidrógeno identificado como H3 en la Figura 6, dejando las demás para un estudio futuro más detallado el radical resultante de dicha abstracción puede verse en la Figura 7, como es fácil notar los átomos del radical tienen posiciones ligeramente diferentes a las del 1-penten-3-ol; en ese sentido una comparación entre las distancias interatómicas antes y después de la abstracción puede verse en la Figura 8.
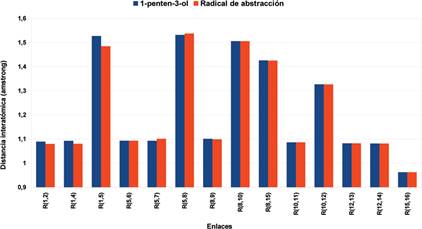
Figura 8 Distancias interatómicas del 1-penten-3-ol y el radical generado por la abstracción del hidrógeno H3 con un nivel de cálculo DFT/B3LYP/aug-cc-pVTZ.
Se puede apreciar que las distancias interatómicas entre el carbono y los hidrógenos del metilo del 1-penten-3-ol disminuyen al ser abstraído el hidrógeno H3, las disminuciones son del 0,85% y del 1,11%. También es interesante notar que las distancias entre el los carbonos 5 y 8 y entre el carbono 5 y el hidrógeno 7 (Figura 6) han aumentado en 0,75% y 0,38% respectivamente, siendo prácticamente las únicas que han cambiado en ese sentido. Las energías relacionadas con el radical obtenido se pueden ver en la Tabla 4, en ella la energía a base infinita fue determinada con la ecuación (2), también se reportan las energías con la corrección del punto cero y al comparar las mismas con las extrapolaciones a base infinita se puede notar que las últimas arrojan valores de energía menores.
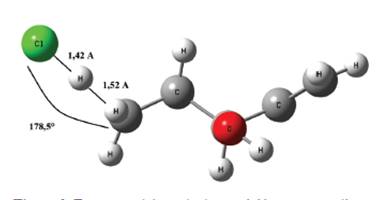
El sistema en el estado de transición para este proceso de abstracción se muestra en la Figura 9, el mismo fue obtenido por la optimización de la geometría a nivel B3LYP/aug-cc-pVTZ se puede apreciar que la distancia entre el hidrógeno y el cloro 1,42 Å mientras que la del hidrógeno al carbono es de 1,52 Å, la cual ha aumentado en comparación a la que mantenía en la estructura del alcohol que era de tan solo 1,09 Å, por otro lado es interesante notar que el ángulo CHCl es muy próximo a 180°, asimismo se reporta un número de onda asociada a la frecuencia imaginaria de 383,25i cm-1.
Las energías relacionadas con este estado de transición son reportadas en la Tabla 5.
Tabla 5. Energías (Eh) de la estructura del estado de transición para la abstracción del hidrógeno 3.
Para esta reacción se realizó además el estudio del camino de reacción, el cual permitió construir el perfil de energía de la misma, este se muestra en la Figura 10.

Finalmente y en cuanto a lo que corresponde al estudio de abstracción es importante hacer notar que desde el punto de vista energético, las abstracciones correspondientes a los hidrógenos del CH3 son equivalentes, es decir las energías de los radicales resultantes son iguales.
Reacción de adición
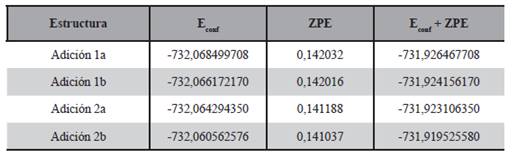
En cuanto a las adiciones posibles al doble enlace del carbono se analizaron cuatro de ellas, las estructuras finales de estas adiciones, optimizadas al nivel B3LYP/aug-cc-pVTZ son mostradas en la Figura 11. Estas adiciones fueron denominadas 1a y 1b cuando la adición de cloro al doble enlace del carbono se lleva a cabo al carbono 1 de la estructura y en un plano cercano al oxígeno del OH (lado a) o en el plano más alejado (lado b), el mismo criterio se tuvo en cuenta a las adiciones 2a y 2b que corresponden al carbono 2 del 1-penten-3-ol, en concordancia con la denominación dada por Rodríguez y colaboradores (2010). Las energías de estas moléculas calculadas mediante la optimización de sus geometrías al nivel B3LYP/aug-cc-pVTZ se muestran en la Tabla 6.

La adición 2b de la Figura 11 fue estudiada con detalle concluyendo que la adición se da con una longitud de enlace entre el carbono y el cloro de aproximadamente 1,92 Å. Asimismo fue posible notar que las longitudes de enlace del 1-penten- 3-ol con relación a las del radical obtenido por la presente adición del cloro, de los átomos cercanos al cloro adicionado, aumentaron. También se pue- de ver en la Tabla 6 que el radical obtenido por adición de menor energía es el correspondiente al proceso 1a, tanto para las energías conformacionales como para aquellas con corrección del punto cero.
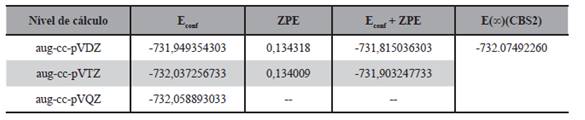
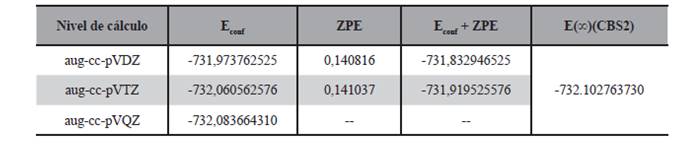
Desde el punto de vista de la energía, los valores calculados para el radical obtenido por la adición del cloro al doble enlace se reportan en la Tabla 7, en la misma se puede observar las energías conformacionales, las del punto cero, la corrección de la energía por adición de la energía del punto cero y la extrapolación a base infinita dada por el modelo CBS2 utilizando la ecuación (2).
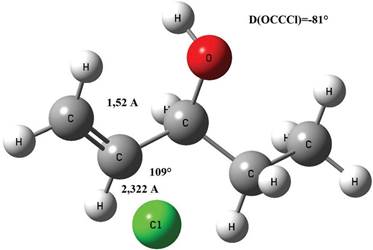
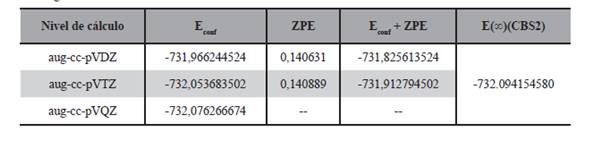
El estado de transición asociado a este proceso de adición se reporta en la Figura 12, el mismo fue obtenido por optimización de la geometría a nivel B3LYP/aug-cc-pVTZ. Algunos datos relevantes pueden verse en dicha Figura, en ella se puede apreciar que la longitud del enlace CC es de 1,512 Å, en la literatura consultada (Rodríguez et al., 2010) se reporta 1,511 Å, la distancia entre el Cl y el C es de 2,322 Å mientras que en el mismo trabajo se reporta 2,324 Å, el ángulo entre los dos carbonos y el Cl es de 109° mientras que se reporta 108° y finalmente el ángulo diedro de -81° en el mismo artículo se reporta -84°, la frecuencia imaginaria asociada al estado de transición es 186i cm-1 la misma en el trabajo arriba reportado (Rodríguez et al., 2010) es de 410i cm-1, un valor evidentemente diferente al hallado en este trabajo, es posible que esta diferencia se deba a los distintos niveles de cálculo utilizados. Por otro lado también se puede decir que la mayor parte de las distancias interatómicas son menores en el estado de transición.
En cuanto a las energías involucradas en este estado de transición, las mismas son reportadas en la Tabla 8.
Para esta reacción se realizó además el estudio del camino de reacción, el cual permitió construir el perfil de energía de la misma, este se muestra en la Figura 13, de este perfil se puede deducir la existencia de un mínimo en la SEP, el mismo se encuentra en un nivel energético aproximadamente 17,4 kcal/mol menor que el nivel en el cual se encuentran los reactivos. Entre este mínimo y el estado de transición hay una barrera energética aproximadamente igual a 7,1 kcal/mol y como se puede apreciar el estado de transición es más energético que el complejo pre-reactivo, en síntesis es necesario agregar 7,1 kcal/mol.
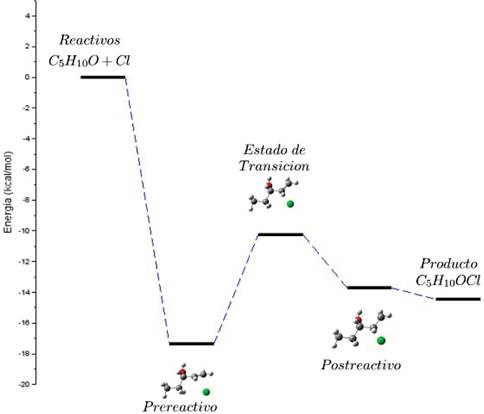
Por otro lado se puede apreciar también que el estado de transición se encuentra en un nivel energético 3,5 kcal/mol superior a lo que se denominó complejo post-reactivo y 4,2 kcal/mol por encima del nivel de energía de los productos.
Es importante notar que existe una barrera de activación entre el estado de transición y los reactivos de -10,2 kcal/mol, lo cual hace que la reacción sea favorable, de igual manera es posible notar que el producto de la reacción es 14,4 kcal/mol menos energético que los reactivos, lo cual haría que la reacción sea exoenergética.
Conclusiones
El 1-penten-3-ol fue modelado tanto a nivel B3LYP/aug-cc-pVDZ como B3LYP/aug-cc- pVTZ, su geometría fue optimizada y se realizaron detallados análisis de la misma Igualmente fue calculada su energía de conformación, fue determinada su energía de punto cero y extrapolado el cálculo de energía a base infinita con cinco modelos diferentes de extrapolación, llegando a la conclusión que en todos los casos el modelo que reducía más la energía era el modelo potencial dado por la ecuación 2. En cuanto a los cálculos de energía también se pudo comprobar que las energías determinadas van disminuyendo conforme aumenta el tamaño de la base utilizada, en la medida de lo posible los cálculos determinados fueron comparados con resultados experimentales y/o resultados teóricos, estando los mismos en buena concordancia.
También fueron estudiados los distintos rotameros del COV analizado, hallándose 6 posibles, cuatro más de los reportados en la bibliografía consultada.
Asimismo, fue modelada completamente una abstracción del hidrógeno y cuatro adiciones al doble enlace del carbono, y fue elegida una adición al doble enlace para profundizar su estudio, llegándose para ellas a determinar estados de transición y perfiles de energía tanto para la abstracción realizada como para la adición elegida.