









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkINTRODUCCIÓN
La actividad económica y el crecimiento de la sociedad en las ciudades tienen como consecuencias la aparición de nuevos contaminantes denominados “contaminantes emergentes” (Benítez-Villalba et al., 2017; Cunningham et al., 2006; Calamari, 1995; Richardson & Bowron, 1985; Doughton & Ternes, 1999). Dentro de este grupo de contaminantes, se incluye una vasta variedad de productos farmacéuticos, antimicrobianos y numerosos compuestos químicos con actividad como disruptores endocrinos. Muchos de estos compuestos van a las aguas residuales urbanas e industriales, pudiendo ser degradados y eliminados durante el tratamiento de las mismas. Sin embargo, se ha demostrado que los procedimientos de depuración en las estaciones depuradoras de aguas residuales (EDARs) no son completamente efectivos, ya que estas sustancias permanecen en los efluentes tratados, pudiendo reingresar al medio ambiente convirtiéndose en un serio peligro para los ecosistemas11-13. (Fent et al., 2006; Jemba, 2006).
Generalmente, los lodos de depuradoras obtenido como subproducto en los EDARs son sometidos a tratamientos de compostaje, que presenta numerosas ventajas tales como la desinfección de los lodos, la eliminación por vía microbiana de algunos contaminantes, y la obtención de un sustrato rico en materia orgánica y nutrientes, apto para el enmendado de suelos agrícolas. Actualmente, se conoce muy poco acerca de la capacidad que tiene este proceso en la eliminación definitiva de estos contaminantes emergentes. Tampoco se tienen evidencias de que parámetros o factores favorecen la degradación de estas sustancias durante este proceso. Por otro lado, se desconoce de manera exacta del efecto que produce el enmendado de los suelos con lodoso compost sobre los contaminantes presentes, así como el riesgo real al que están expuestos los seres humanos, ya que estas sustancias pueden transferirse, por ejemplo, a las aguas subterráneas17,18. (Benítez‐Villalba et al., 2013; Jemba, 2006; Fent et al., 2006). En la actualidad no se cuenta aún con evidencia suficiente, que la presencia de estas sustancias en los suelos puede representar un riesgo para la salud humana y los ecosistemas, ya que al encontrarse adsorbidos, los microorganismos del suelo no pueden biodegradarlos satisfactoriamente, convirtiéndose en sustancias bioacumulables, persistentes y tóxicos; existiendo la posibilidad de ser transferidos a las cosechas, y de ingresar en la cadena alimentaria de los seres vivos (Khetan & Collins, 2007; Kummerer, 2004; Bush, 1997).
En este sentido, el desarrollo de nuevas técnicas analíticas, permite determinar concentraciones de compuestos a nivel ultra traza (μg-ng L-1), en matrices complejas como son las medioambientales (Jemba, 2006; Petrovic et al., 2005; Díaz-Cruz et al., 2006). Entre estos compuestos se encuentran las benzofenonas que son cetonas aromáticas muy usadas en numerosas aplicaciones como, perfumería, reactivos en síntesis orgánicas y en productos como jabones y protectores solares. Son sustancias blancas cristalinas, insolubles en agua y con olor a rosa (Sánchez-Brunete et al., 2011; Kotnik et al., 2014; Benítez‐Villalba et al., 2013; European Commission, 1996). En la Figura 1 de observa las benzofenonas estudiadas en este trabajo de investigación.
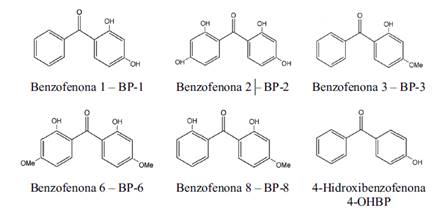
Existe escasa información sobre la determinación de benzofenonas en suelos, se ha encontrado trabajos en sedimentos utilizando la técnica de extracción por líquidos presurizados (PLE) y la técnica de cromatografía gaseosa acoplada con espectrometría de masas, mientras que el estudio de estos contaminantes en causes hídricos son más numerosas (Jeon et al., 2013; Sánchez-Brunete et al., 2011). Estos contaminantes ingresan a los ecosistemas terrestres a través de lodos de depura- dora tratados o compostados que se utilizan como fertilizantes en el enmendado de suelos agrícolas (Benítez‐Villalba et al., 2013; Jemba, 2006; Fent et al., 2006).
Anualmente, se liberan al agua miles de toneladas de contaminantes emergentes, estimándose en unos 6 millones el número de sustancias disponibles comercialmente y su uso se está incrementando en un 3-4 % en peso cada año (Kummerer, 2003). Por ello, la nueva preocupación medioambiental y de salud pública ha sugerido a la comunidad científica, que se debe hacer gran esfuerzo para investigar este problema e intentar paliar en la medida de lo posible sus efectos (Khetan & Collins, 2007; Kummerer, 2004; Bush, 1997). Este trabajo de investigación se ha realizado con el objetivo de desarrollar, optimizar y validar un método analítico sensible con el fin de poder cuantificar una familia de benzofenonas en muestras de suelos contaminados, y que pueda ser aplicado en el futuro para el monitoreo de estas especies químicas en muestras de suelos agrícolas o futuros estudios cinéticos de degradación o biodegradación de estos compuestos en la matriz mencionada para entender su comportamiento medioambiental.
MATERIALES Y MÉTODOS
Productos químicos Reactivos
Se utilizaron reactivos de grado analítico. Los patrones: Benzofenona 1 (BP-1), Benzofenona 2 (BP-2), Benzofenona 3 (BP-3), 4-hidroxibenzofenona (4-OHBP), Benzofenona 6 (BP-6), Benzofenona 8 (BP-8) y Benzofenona deuterado (BP-d-10) fueron suministrado por la casa comercial Sigma-Aldrich (St. Louis, MO, USA). Las soluciones de los ana litos (200 mg.mL-1) se prepararon mensualmente y se almacenaron a -20 °C. Las soluciones estándar, mezclas de estos compuestos junto con el patrón interno se prepararon con metanol o en la fase móvil inmediatamente antes de su uso. Estos estándares se almacenaron en botellas de vidrio oscuro. El metanol y acetonitrilo que se utilizaron para la preparación de la fase móvil fueron de grado LC- MS suministradas por Fluka (St. Louis, MO, USA). El amoníaco y acetato de etilo se adquirieron de Merck (Darmstadt, Alemania). El agua (18,2 MΩ cm) se purificó por un sistema Millipore Milli-Q (Bedford, MA, USA).
Instrumentación y software
Para la extracción de los analitos de la matriz estudiada se utilizó un equipo de Liquido Presurizados Dionex ASER 200 (Sunnyvale, CA, USA) equipado con un controlador de solvente para la extracción. Un cromatógrafo Waters Acquity UPLCTM acoplado a espectrómetro de masas triple cuadrupolo Waters H-Class-Xevo TQSTM con una fuente de ionización por electroespray Z-sprayTM ESI para la detección de los analitos. La separación de los compuestos se obtuvo utilizando una columna ACQUITY UPLC BEHTM C (1,7 µm; 2,1 mm × 50 mm) (Waters). El control del instrumento, la detección de picos y la integración se llevaron a cabo utilizando el software MassLynx (versión 4.1). Se utilizaron una centrífuga Hettich Universal 32 (Tuttlingen, Alemania) y un horno Memmert (Schwabach, Alemania). Se utilizó un software Statgraphics Plus versión 5.0 (Manugistics, Rockville, MD, USA, 2000) para el tratamiento estadístico de los datos. Paquetes Microsoft® Office: Word®, Excel® y PowerPoint® 2007.
Optimización del equipo UPLC-MS/MS
MS/MS: El método de detección del espectrómetro de masas se optimizó mediante infusiones directa individuales de cada compuesto para optimizar la respuesta del ión precursor. Para una mayor sensibilidad y selectividad, se realizaron los análisis de las masas en modo SRM y los parámetros MS/MS, se optimizaron individualmente para cada benzofenonas mediante la infusión directa de una solución estándar de 0,5 mg.mL-1 disuelta en metanol en el espectrómetro de masas en modo combinado, usando el aducto [M-H]+ como el ión precursor para las mayorías de los analitos y para el BP-2 se operó en modo negativo [M-H]-. También se optimizaron el voltaje capilar, la fuente y la temperatura de de solvatación, el flujo de gas de cono, el flujo de gas de solvatación, el flujo de gas de colisión y el flujo de gas del nebulizador (algunos de estos parámetros afectan fuera de la sonda, pero su optimización se realizó junto con los parámetros adecuados). La optimización se desarrolló inyectando estándares de analitos individuales a un flujo fijo y sin columna cromatográfico.
UPLC: Para la optimización del método cromatográfico, se empleó una disolución metanólica de los analitos a una concentración de 100 μg L-1. Se estudiaron las principales variables que afectan a la separación cromatográfica y la intensidad de la señal, fueron evaluadas las fases estacionarias dadas las características de los analitos seleccionados. La fase móvil, se ensayaron con agua desionizada, metanol y acetonitrilo, con y sin aditivos. Los aditivos estudiados fueron el acetato de amonio (20 mM, pH 9.2) y la solución de amoniaco al 0,025% v/v que se agregaron como modificadores de la fase móvil. Se ensayaron varias gradientes una vez definida la fase móvil óptima. También se evaluaron el efecto del caudal, la temperatura de la columna y los volúmenes de inyección.
Optimización del proceso de extracción Recolección y almacenamiento de las muestras
Se recolectaron muestras de suelo emendadas con compost elaborados con lodos provenientes de una estación depuradora de aguas residuales EDARs, de un campo experimental situada en la Huerta de Santa María en la Vega de Granada - España en el municipio de Belicena a 5 km de la capital y de coordenadas 37° 11’09.50’’ N; 3° 41’39.85’’ O. En el suelo agrícola no se ha utilizado ningún tipo de plaguicidas en los últimos 10 años con el objetivo de no alterar la microbiota del suelo (Kinney, C. A.et al., 2006) y el compost utilizado en el experimento no contenían benzofenonas ya que fueron previa- mente analizadas en el laboratorio. Se recolectaron las muestras de la parte superior del suelo (0 - 30 cm) utilizando un muestreador de 2 cm de calado y una barrena helicoidal tipo EIJKELKAMP . Cada una de las muestras de suelo tomadas se sometió a las siguientes etapas para asegurar su correcto análisis en el laboratorio. Se adicionó formaldehido 1 % (m/v), para evitar el crecimiento microbiano en las muestras. Se secaron y se trató las muestras en bandejas de plástico y al sol a temperatura ambiente. Una vez secas se molturaron con un mortero de hierro y se tamizaron con ayuda de una malla de 2 mm. Posteriormente se introdujeron en una bolsa de plástico y se llevaron al laboratorio donde permanecieron a una temperatura de 4 °C hasta su análisis. Una vez en el laboratorio, las muestras de suelo fueron analizadas.
Preparación de muestras fortificadas
Debido a la ausencia de un material de referencia certificado, se agregaron a 0.5 g de muestras de suelo, 1 mL de una solución estándar metanólica que contenía los analitos. Este volumen hace que los analitos entren en contacto con toda la muestra. Para alcanzar el equilibrio de sorción y permitir la evaporación completa de las muestras, las mezclas se agitaron durante 2 minutos en un vortex y luego se dejaron reposar 24 horas a temperatura ambiente en la oscuridad antes del análisis.
Optimización de la extracción por líquidos presurizados PLE
La extracción de los analitos se realizó por el método de Extracción por Líquidos Presurizados (PLE) (Canosa et al., 2007), se llevó a cabo utilizando un Equipo Dionex, modelo ASE 200. Se optimizaron los parámetros más influyentes en el proceso PLE. En todos los casos, la cantidad de muestra suelo agrícola empleada fue de 0.5 g. Los parámetros optimizados fueron el, disolvente de extracción, tiempo de extracción, temperatura de extracción y la presión. El número de ciclos de extracción realizados a lo largo de todo el proceso de optimización de variables fue de 1, ya que previamente se demostró que el uso de más ciclos no repercutía en una mejora notable de la recuperación, pero aumentaba de forma innecesaria el consumo de disolvente y el tiempo de extracción. Se utilizaron las muestras enriquecidas con 500 ng g-1 de cada compuesto en estudio para las optimizaciones. Las condiciones iniciales fueron las siguientes: temperatura, 140 °C; presión, 1000 psi; tiempo de extracción estática, 25 mini; 1 ciclo; tiempo de purga 90 s, y 30% de volumen de descarga. Se utilizó metanol como disolvente de extracción. El volumen de extracción final fue aproximadamente 7 mL. El extracto orgánico se transfirió a un tubo de centrifuga y se centrifugó a 5000 r.p.m. (3634 x g) durante 30 minutos. El sobrenadante se evaporó a sequedad bajo corriente de nitrógeno. Se disolvió con 1 mL de fase móvil inicial cromatográfica y se agitó en vortex durante unos segundos. No se observó una pérdida significativa de analitos durante este procedimiento. El extracto final se filtró a través de filtro de nylon de 0,45 µm y se transfirió a un vial cromatográfico de vidrio listo para su inyección en el UPLC-MS/MS.
RESULTADOS Y DISCUSIÓN
Espectrómetro de masas MS/MS:
El método de detección MS/MS se optimizó mediante infusiones individuales de cada compuesto para optimizar la respuesta del ión precursor, [M - H]+ para la mayoría de los analitos y [M - H]- para el BP-2. Se monitorizó la respuesta de dos productos de fragmentación iónicos, uno se utilizó para la cuantificación y la otra para la identificación o confirmación del analito. Para la cuantificación se seleccionó la transición más abundante por obtener la máxima sensibilidad. El voltaje del cono (voltaje de desolvatación para romper el disolvente - aducto de analito) se optimizó para cada ión precursor, y la energía de colisión también se optimizó para cada transición para obtener la máxima sensibilidad. La resolución seleccionada para ambos cuádruplos fue de 0.7 Da. Una vez obtenidas las condiciones para todos los analitos, se llevó a cabo una optimización de la sonda del electrospray. Los parámetros óptimos ensayados fueron: voltaje capilar ESI (+), 0,60 kV; Voltaje de capilar ESI (-), 1,8 kV temperatura de la fuente, 150 °C; temperatura de desolvatación, 500 °C; flujo de gas cónico, 150 L/h; caudal de gas de desolvatación, 800 L/h; flujo de gas de colisión, 0.15 mL/min, y flujo de gas del nebulizador, 7.0 bar. Se utilizó nitrógeno (≥ 99.995%) como gas de cono y desolvatación, y argón (99.999%) como gas de colisión. Después de seleccionar los iones precursores, los iones del producto se obtuvieron con una combinación de energías de colisión y voltajes de cono. Se seleccionaron las dos transiciones SRM más sensibles (una utilizada para la cuantificación y la otra para la confirmación). Para la cuantificación, se eligió la transición más abundante por obtener la máxima sensibilidad. El tiempo de espera para cada transición fue de 25 ms, y el retardo entre exploraciones se estableció en 3 ms. Se monitorizó la res- puesta de dos productos de fragmentación iónicos, uno se utilizó para la cuantificación y la otra para la identificación o confirmación del analito. Las energías de colisión y los voltajes de cono óptimos seleccionados para cada transición y los tiempos de retención se muestran en la Tabla 1. Solo se seleccionó una transición para el estándar interno porque la sustancia seleccionada es un compuesto marcado isotópicamente que es poco probable que se encuentre en muestras ambientales.
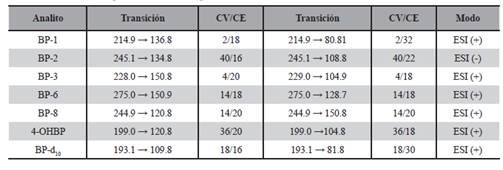
Toda la optimización del MS/MS se desarrolló infusionando estándares de analitos individuales a un flujo fijo y sin columna cromatográfico. Todos estos parámetros optimizados ayudaron a mejorar y obtener una sensibilidad óptima. Una vez que se establecieron todas las condiciones estudiadas se establecieron los tiempos de retención en el método cromatográfico.
Cromatógrafo de líquidos UPLC:
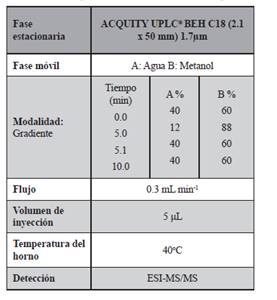
Dadas las características de los analitos seleccionados, en el desarrollo de la metodología propuesta se ha empleado la cromatografía en fase reversa (fase estacionaria menos polar que la fase móvil). Se seleccionó una columna con relleno C18 ACQUITY UPLC BEHTM (2,1 mm × 50 mm) y 1.7 µm de tamaño de partícula. Para la fase móvil, se evaluaron el metanol y acetonitrilo como fase móvil orgánicas (disolvente B) siendo el metanol la utilizada para la fase móvil por dar mejor señal analítica. Se evaluó el agua desionizada (disolvente A) con y sin aditivos. De los aditivos seleccionados para el estudio acetato de amonio (20 mM, pH 9.2) y la solución de amoniaco al 0,025% v/v que se agregaron como modificadores de la fase móvil, la sensibilidad para la detección de los analitos mejoró significativa- mente en condiciones alcalinas porque la ionización de compuestos con grupos fenólicos (grupos ácidos) se logra mejor en estas condiciones. Los resultados óptimos se obtuvieron con amoníaco, que también se añadió al metanol (disolvente B) a la misma concentración. De la comparación de las interfaces ESI y APCI con el aditivo óptimo en la fase móvil, el ESI en modo positivo proporcionó la ionización óptima para la mayoría de los analitos y también se observó menos supresión de la ionización debido a los efectos de la matriz. Las condiciones de gradientes óptimo fueron: fase móvil inicial, 60% (B), que se incrementó linealmente a 100% (B) en 4.0 min y se mantuvo durante 2.5 min para limpiar la columna. Finalmente, volvió al 60% en 0.1 min y se mantuvo 3.4 min para equilibrar la columna. El tiempo total de ejecución fue de 8 min. Para el estudio del efecto caudal se ensayaron velocidades de flujo de 0.050 a 0.350 mL/min y este parámetro mejoró significativamente la resolución, la forma del pico, la intensidad de la respuesta y los tiempos de retención. El caudal óptimo seleccionado fue de 0.300 mL/min que mejoró la resolución de los picos cromatográficos y los tiempos de retención. Se ensayaron temperaturas de la columna de 30 °C a 50°C y la temperatura óptima seleccionada fue de 40 °C que mostró en todos los casos una buena forma de pico. Finalmente se evaluaron los volúmenes de inyección desde 2 a 10 µL y se seleccionó 5 µL como óptimo (ver valores óptimos en la Tabla 2).
Optimización de la extracción por líquidos presurizados PLE
En la optimización del disolvente de extracción se empleó una metodología univariante. Se ensayaron varios disolventes teniendo en cuenta el rango de polaridad que cubren estos compuestos: metanol, Acetonitrilo y acetato de etilo. Las mejores recuperaciones, para la mayor parte de los analitos, se obtuvieron cuando se utilizó metanol como disolvente de extracción (Figura 2). Por tanto, este disolvente fue elegido para continuar la optimización de las variables de extracción.
Para la optimización de las variables (tiempo, temperatura y presión), se llevó a cabo un estudio de las tres de manera conjunta mediante un diseño experimental de tipo Doehlert. Este diseño describe un dominio experimental esférico, ofreciendo una distribución uniforme de los experimentos en un espacio tridimensional cuboctaedro, de modo que 12 experimentos son equidistantes de un experimento central, describiendo una esfera de radio uno (Sautour et al., 2001). El ensayo consistió de quince experimentos (tres de ellos puntos centrales), siendo seleccionada como variable de respuesta la suma de las áreas de respuesta de la familia de los compuestos objeto de estudio. Los máximos y mínimos de cada factor experimental, se establecieron de acuerdo a las posibilidades del sistema instrumental. Es decir, para el tiempo de extracción, el valor mínimo fue de 5 min y el máximo de 25 min; para la temperatura, el mínimo fue de 80 °C y el máximo de 140 °C y para la presión el mínimo fue de 500 psi y el máximo de 1500 psi. El procedimiento se- guido para la optimización de estas variables fue el siguiente: (i) Se pesaron 0.5 g de muestra sólida en una celda de extracción. (ii) Se fortifico la muestra con una disolución metanólica conteniendo todos los analitos de estudio. (iii) Se agito durante 2 min en un vortex y se dejó reposar durante al menos 24 h a temperatura ambiente, con el objetivo de alcanzar un equilibrio entre los analitos y la matriz. Una vez transcurrido este tiempo, se llevó a cabo la extracción de las muestras.
La evaluación de los resultados del diseño experimental se basó en el estudio de ciertos parámetros como el grafico de Pareto, el análisis de la varianza, el grafico de la superficie de respuesta y el óptimo calculado. El primer parámetro estudiado fue el grafico de Pareto. A continuación, se muestran los gráficos obtenidos en este estudio:
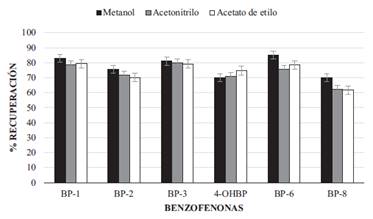
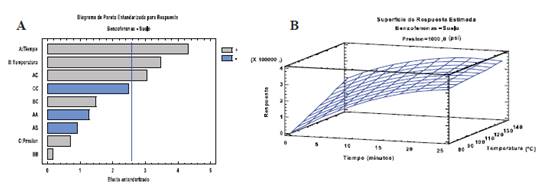
Figura 3 A) Gráfico de Pareto B) Gráfica de superficie de respuesta de la familia de benzofenonas en muestra de suelo agrícola estudiada.
El resultado obtenido a partir de este grafico de Pareto Figura 3. A) para las benzofenonas de- muestra que las dos variables estudiadas, tiempo y temperatura, tienen un efecto positivo en el proceso de extracción y ambas son significativas para esta familia de compuesto, mientras que la presión es positiva, pero no significativo.
La siguiente etapa fue realizar un ANOVA, determinándose el valor de % R2, para así conocer la idoneidad del diseño aplicado. El valor calculado fue de 93.17% demostrando un buen ajuste de los datos al modelo propuesto.
Finalmente se procedió al estudio de las superficies de respuesta Figura. 3 B) obtenidas con objeto de determinar de manera gráfica cuál es el óptimo de las condiciones propuestas. El óptimo fue calculado maximizando la ecuación de la curva que da lugar a la representación de la superficie de respuesta. Los resultados obtenidos demuestran que el tiempo y la temperatura producen un efecto positivo en el proceso de extracción. Se fijó 25 min para el tiempo de extracción y 140 oC para la temperatura. Estos valores son próximos a los óptimos obtenidos para la mayoría de los analitos en esta matriz. Para la presión el óptimo obtenido fue de 1500 psi.
A continuación, se presenta un resumen de los valores óptimos y se detalla el procedimiento final de extracción PLE aplicado a la matriz estudiada cuando se empleó esta técnica de extracción. Se pesan 0.5 g de muestra en la célula metálica, y se extrae en el equipo fijando las condiciones mostradas en la Tabla 3.
El extracto orgánico obtenido se trasfirió a un tubo de centrifuga y se centrifugó durante 30 min a 5000 r.p.m. (3634 x g). El sobrenadante fue recogido sobre un vial de vidrio y se evaporó a sequedad bajo corriente de N2. A continuación, se re disolvió con 1 mL de la fase móvil inicial cromatográfica, y se agitó en vortex durante unos segundos. El extracto final, se filtró a través de filtro de nylon de 0.45 µm y se transfirió a un vial cromatográfico de vidrio, quedando listo para su inyección en el LC-MS/MS.
Validación del método analítico
El método analítico se validó en términos de linealidad, selectividad, sensibilidad y precisión (veracidad y reproducibilidad). Para establecer la metodología analítica en la muestra medioambiental seleccionada se procedió del siguiente modo:
se realizó un calibrado externo con patrones en disolvente para cada uno de los analitos en estudio.
Tabla 3 Variables optimizadas y las condiciones de extracción final para el procedimiento por PLE en suelos.
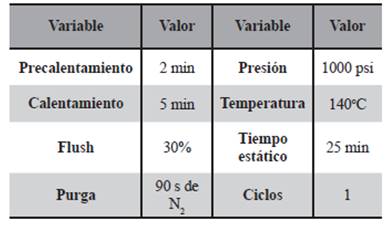
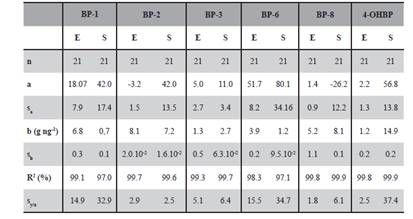
Tabla 4 Parámetros del calibrado externo benzofenonas (E) y del calibrado en suelos (S). n = puntos de calibración, a = intercepto, sa = desviación estándar del intercepto, b = pendiente, sb = desviación estándar de la pendiente, R2 (%) = Coefiente de correlación, Sy/x = desviación estándar de la regresión.
se realizó un calibrado en matriz para cada uno de los analitos. (iii) se compararon las pendientes de ambos calibrados mediante un test estadístico de t-student. El patrón interno seleccionado de acuerdo a las propiedades físico-químicas para realizar los calibrados fue la Benzofenona deuterado (BP-d-10), utilizando en todos los casos la relación área del pico del analito/área del pico del patrón interno y la concentración de analito. Los requisitos de la validación del método analítico se establecieron de la siguiente manera: (a) Linealidad, el coeficiente de determinación (R2) debe ser igual o mayor que 0.990 y la desviación residual máxima debe ser menor al 25%. (b) La precisión, expresada como desviación estándar relativa DER (precisión entre días) debe ser ≤ 30%. (c), la veracidad, expresada como recuperación media, debe estar en el rango 70 a 120%. (d) El LC (límite de cuantificación) debe cumplir con los requisitos (b) y (c). Estos requisitos se ajustan a la Decisión 2002/657 / CE de la Comisión.
En el calibrado en disolvente se prepararon patrones conteniendo cantidades creciente de los analitos (3 réplicas por nivel) 7 niveles de concentración y una concentración constante del patrón interno (0.6 μg kg-1). Para el calibrado en matriz también se preparó una curva de 7 niveles de concentración (con 3 réplicas de cada nivel) mediante la adición de una solución patrón mezcla de todos los analitos en metanol. Se añadió el patrón interno para obtener la concentración del extracto final BP-d10 (0.6 μg kg-1) y se extrajo según el procedimiento previamente optimizado en la técnica PLE. En la Tabla 4 se muestran los resultados obtenidos de los parámetros analíticos para el calibrado externo y en matriz.
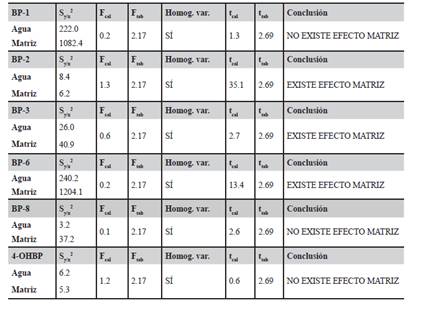
Se evaluó el efecto matriz comparando los dos calibrados, externo y en matriz para determinar si existen diferencias significativas entre ambos, en la Tabla 5, se presentan los resultados del test estadístico de la comparación del calibrado externo y en matriz evaluando así el efecto matriz.
Al evaluar los resultados obtenidos para cada uno de los analitos estudiados en las muestras de suelo se puede observar que, en la mayor parte de los casos, existen diferencias significativas entre las pendientes en el calibrado externo y el calibrado en suelo en todos los casos, por lo que se puede decir que existe efecto matriz. Con esto se confirma que la correcta cuantificación de los analitos en la muestra de suelo requiere el uso de las curvas de calibrados basadas en matriz. Dos aspectos fundamentales se examinaron en esta validación de método analítico que se realizó cuando el analito se encuentra presente en la muestra: el Límite de Detección (LD) y el Límite de Cuantificación (LC) cuyos resultados se encuentran en la Tabla 6. En la misma, también se detallan los valores del Rango Dinámico Lineal (RDL) propuestos para cada analito en la matriz estudiada. Los valores obtenidos de %R2 fueron próximos al 100 % en todos los casos, demostrando que existe linealidad en el intervalo estudiado para todos los analitos en la matriz de estudio; además de la sensibilidad del método analítico o sensibilidad de la calibración que se ha medido mediante la relación entre la señal instrumental y la concentración del analito del punto 2 del rango lineal, y viene dada por la pendiente de la recta de calibrado.
Exactitud del Método: Precisión y Veracidad
Para establecer la exactitud del método propuesto, se ha estudiado la veracidad del mismo en términos de recuperación con muestras dopadas y su precisión en términos de variabilidad inter e intradía. Uno de los mayores problemas que se encuentran a la hora de estudiar la exactitud de un método, en términos de veracidad, especialmente en matrices ambientales poco investigadas, es la ausencia de materiales certificados. Por este motivo, en este trabajo, se ha llevado a cabo un estudio de recuperación con muestras dopadas, a un nivel de concentración cercano al límite de cuantificación, empleando para ello muestras limpias de los compuestos o cuyas concentraciones están por debajo de los límites de detección del método. Las muestras fueron analizadas empleando el procedimiento operatorio descrito a lo largo de este trabajo y la concentración de cada analito en la muestra se determinó por interpolación a la curva de calibración dentro del rango dinámico lineal. Los valores de recuperación se obtuvieron por comparación con las cantidades de analito conocidas que fueron añadidas previamente a las muestras.
Tabla 6 Términos de validación del método analítico: Límite de Detección (LD), Límite de Cuantificación (LC), Rango lineal dinámico, Sensibilidad analítica, Porcentajes de recuperación (%R) Precisión (%RSD).

Por su parte, para evaluar la exactitud del método en términos de precisión, se realizaron diversas réplicas de los análisis llevados a cabo en el estudio de veracidad, durante un mismo día y durante diez días consecutivos. En las tablas siguientes se muestran las recuperaciones de los ensayos realizados para cada analito durante la validación (%R) y en los valores obtenidos para la precisión, expresada como desviación estándar relativa (%DER).
Los promedios de los porcentajes de recuperación para las muestras de suelo fueron próximos al 99 % (entre el 98 % para el BP-8 y el 101 % para BP-3 en la matriz estudiada), lo que nos lleva a deducir que el procedimiento de extracción es eficiente y el método veraz. Por otro lado, los valores obtenidos para el DER fueron menores al 4% en todos los casos. Estos datos se encuentran dentro de límites aceptables para cumplir los requisitos establecidos en la guía de validación empleada, según la cual se consideran aceptables valores iguales o inferiores al 15% en general, y al 20% en la zona próxima al límite de detección del método, por lo que se pudo concluir que el método propuesto para las muestras de suelo utilizando la técnica de extracción por líquidos presurizados genera resultados próximos entre sí, cumpliendo de este modo los requisitos de precisión. Se trata por tanto de un método veraz, preciso y por tanto exacto.
A continuación, se muestran los cromatogramas de suelo dopadas utilizando el método optimizado por líquidos presurizados (Figura 4).
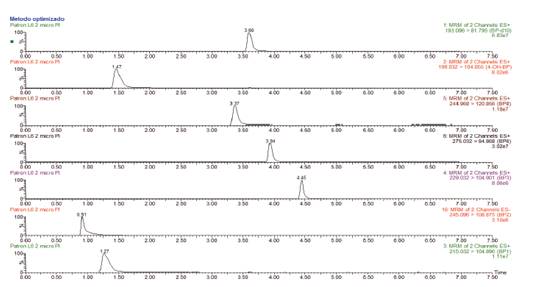
CONCLUSIÓN
Este trabajo de investigación propone la validación de un método analítico, basado en la extracción por líquidos presurizados PLE seguido por el análisis por cromatografía de líquidos acoplada a un espectrómetro de masas en tándem, para la determinación y cuantificación de una familia de benzofenonas en muestras de suelos. Las benzofenonas son ampliamente utilizadas en perfumería, jabones y protectores solares entre otros, y se consideran productos químicos de alteración endocrina. Aunque los extractos mostraron efectos en la matriz para todos los analitos, la optimización de extracción por líquidos presurizados, junto con la detección y cuantificación por UPLC-MS/MS permitieron una sensibilidad analítica y un rendimiento analítico del método.
Este método proporcionó un LD bajo (entre 0.05 y 0.28 ng g-1), recuperaciones altas y buena precisión, lo cual es un logro importante en comparación con otros métodos que involucran más procedimientos. La técnica de extracción por líquidos presurizados podemos considerar una técnica de extracción óptima debido a buen rendimiento de extracción, fácil operación, tiempos de análisis corto y el alto grado de automatización, con alta sensibilidad y precisión. La contribución más importante de este trabajo es que se ha desarrollado un método analítico que muestra buenos parámetros analíticos como la precisión y la sensibilidad en la determinación de los compuestos que fueron seleccionados para el estudio del mismo en los suelos agrícolas y que puede ser aplicado en el futuro para el monitoreo de estas especies químicas en muestras de suelos agrícolas o futuros estudios cinéticos de degradación o biodegradación de estos compuestos en suelo agrícola para entender su comportamiento medioambiental.