











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La hemostasia “es el conjunto de sistemas que actúan coordinadamente para mantener la integridad de los vasos sanguíneos y la fluidez de la sangre” 1. Los sistemas encargados de mantener el balance hemostático son; el vascular, el plaquetario, el de la coagulación y el de fibrinólisis, y la alteración de cualquiera de estos puede desencadenar trastornos trombóticos o hemorrágicos 1.
Los trastornos trombóticos o hemorrágicos pueden ser tan graves que se precisa del soporte hospitalario para su tratamiento como en los trastornos trombóticos, a veces inadvertidos como las deficiencias leves de factores o francamente hemorrágicos como las Hemofilias severas.
Estos trastornos son estudiados teniendo en cuenta el esquema clásico de la coagulación (vías intrínseca, extrínseca y común) y son útiles para la orientación del diagnóstico de los síndromes hemorrágicos, a pesar de ser escasamente fiel a la coagulación fisiológica2,3.
En la actualidad, las superficies celulares (plaquetas, células endoteliales, fibroblastos y monocitos) juegan un papel esencial en la coagulación sanguínea4,5.
De hecho dos pruebas coagulativas básicas, como el tiempo de protrombina (TP) y el tiempo de tromboplastina parcial activada (TTPA), son fundamentales para detectar una gran parte de las alteraciones debidas a deficiencias de factores del sistema de la coagulación6, excepto el factor XIII (FXIII) no explorado por estas pruebas.
Con la automatización de la hemostasia que se inicia de a poco en la década del 70, se logró mejorar considerablemente la exactitud (veracidad) y la precisión (reproducibilidad) de las pruebas7, que es importante para asegurar la analítica y así cooperar con el diagnóstico de las coagulopatías.
En nuestro país ningún otro centro asistencial dependiente del Ministerio de Salud Pública y Bienestar Social o privado, tiene la cartera de servicios de Hemostasia que ofrece el Hospital Nacional de Itauguá; las pruebas básicas, el dosaje de los factores desde el I al XII, entre ellos los factores VIII y IX para el diagnóstico de las Hemofilias A y B, Factor de Von Willebrand, los principales inhibidores fisiológicos, orientación hacia los de interferencia y titulación del Anti Factor VIII (aFVIII) uno de los inhibidores más frecuentes entre los adquiridos, además de algunas pruebas que pueden dar información sobre la actividad del sistema fibrinolítico. A todas estas pruebas el paciente puede acceder de modo gratuito.
El 14 de abril de 2015, por resolución N° 167, el MSP y BS se establece el plan estratégico de atención integral a pacientes en condición de hemofilia y designa al Hospital Nacional de Itauguá como Hospital de Referencia, por esa razón el laboratorio de Hemostasia es fundamental para llegar a los diagnósticos de estas enfermedades.
Este trabajo pretende colaborar con la concienciación de la actividad laboratorial para lograr una correcta orientación, diagnóstico y otorgar una buen soporte para el tratamiento de las coagulopatías, ya que muchas de ellas son muy poco frecuentes, como los déficit de factores I, II, V, VII, IX y X10,11.
OBJETIVOS
Describir las coagulopatías hemorrágicas que se registraron en el Departamento de laboratorio del Hospital Nacional de Itauguá, desde julio 2014 hasta diciembre 2015.
MATERIAL Y MÉTODOS
Diseño: observacional, descriptivo, de corte transverso, retrospectivo, de registros de pacientes que se realizaron estudios básicos y específicos de Hemostasia, desde el 01 de julio 2014 hasta el 31 de diciembre del 2015.
Población de estudio: pacientes de ambos sexos, que acuden al Departamento de Laboratorio del Hospital Nacional de Itauguá, para pruebas básicas y específicas de hemostasia.
Criterios de inclusión: pacientes de ambos sexos, de todas las edades que se realizaron estudios básicos de la hemostasia y pruebas específicas, según pedidos derivados de médicos hematólogos del Hospital Nacional y otros centros hospitalarios, al Laboratorio desde julio 2014 hasta diciembre 2015.
Criterios de exclusión: todos los pacientes cuyos resultados fueron normales y los pacientes con plaquetopenia.
Muestreo: fue no probabilístico de casos consecutivos.
Variables: se agruparon en variables sociodemográficas (edad, que fue clasificado en pediatría y adultos, debido a las características de las enfermedades asociadas a la hemostasia y que son atendidas por Hematólogos pediatras y de adultos, sexo, procedencia hospitalaria, ya que se realizaron pruebas a pacientes derivados de médicos hematólogos de otros centros hospitalarios, la residencia de pacientes no fue posible obtener debido a limitaciones del instrumento de medición) y variables relacionadas con las deficiencias de los factores de coagulación (factores I, II, V, VII, VIII, IX, X, XI y XII).
El punto de corte utilizado fue el criterio del Grupo Cooperativo Argentino de Hemostasia y Trombosis (CAHT) (Tabla 1).
El procesamiento en el Laboratorio consistió en tomar las muestras de sangre con citrato de sodio al 3,2 %, recomendado por el Grupo Cooperativo Argentino de Hemostasia y Trombosis y que fueron validados por el laboratorio8,9. Posteriormente, los plasmas fueron procesados en los equipos automatizados de la línea Instrumentation Laboratory ®(IL) Elite Pro ®, con reactivos, controles y calibradores de la misma marca que el equipo. Los controles de calidad utilizados fueron el interno y el externo, pues el laboratorio participa regularmente de un programa de evaluación externa de la calidad de tercera opinión internacional de la marca Randox, cumpliendo lo recomendado por el grupo CAHT8,9.
Para investigar las deficiencias de factores de la vía extrínseca de la coagulación se realizan las pruebas básicas; Tiempo de protrombina (TP), tiempo parcial de tromboplastina activada (TTPA) y Fibrinógeno, al alterarse solo la prueba del TP, se realiza la prueba de mezcla con plasma normal y si corrige, se confirma la deficiencia, por lo que el paso siguiente es la determinación de la prueba específica que es el Factor VII. Para investigar las deficiencias de los factores de la vía intrínseca, se realizan primero las pruebas básicas y si se altera solo la prueba del TTPA, se realiza la prueba de mezcla con plasma normal y si corrige, se confirma la deficiencia, por lo que se realizan las pruebas específicas que son los factores VIII, IX, XI y XII. Para investigar las deficiencias de la vía común se procede a realizar las pruebas básicas, si se alteran las pruebas de TP y TTPA, se realiza la prueba de mezcla y si corrigen ambas pruebas, se confirma la deficiencia, realizándose las pruebas específicas que son los factores I, II, V y X. Si al realizar la prueba de mezcla con plasma normal la prueba del TP o TTPA no corrigen, se sospecha de la presencia de inhibidores de la coagulación que pueden ser específicos o adquiridos, una forma de confirmar la presencia de inhibidores específicos anti factor VIII, es la utilización de la titulación del método Bethesda.
Las variables que se tuvieron en cuenta fueron:
Deficiencias de factores de coagulación: dosaje de factores de coagulación disminuidos
Fibrinógeno llamado factor I: valor de referencia de 1,5 a 4 g/L,
Deficiencia de los factores de la vía extrínseca: constituido por el factor VII. Valor de referencia de 70 a 120%.
Deficiencia de los factores de la vía intrínseca: constituido por los factores: VIII, IX, XI y XII. Valor de referencia de 50 a 150%.
Deficiencia de los factores de la común final: constituido por factores: II, V y X (además del factor I). Valor de referencia 70 a 120%.
Tabla 1 Valores de referencia para los factores de la coagulación
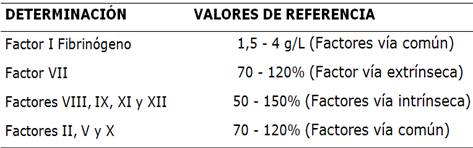
Fuente: Grupo Cooperativo Argentino de Hemostasia y Trombosis
Las deficiencias fueron clasificadas en severas, moderadas y leves, de acuerdo al resultado cuantitativo de los factores en el momento del diagnóstico. Los valores de referencia utilizados para todos los factores de la coagulación, incluido del factor von Willebrand fueron los valores recomendados por el Grupo Cooperativo Argentino de Hemostasia y Trombosis (CAHT), y que fueron validados por el laboratorio, ya que no existen registros de nuestro país (Tabla 2).
Tabla 2 Grado de deficiencia de los factores de la coagulación

Fuente: Grupo Cooperativo Argentino de Hemostasia y Trombosis
En cuanto a los aspectos éticos fueron respetados los criterios de confidencialidad en cuanto a la identidad de los pacientes.
RESULTADOS
Fueron estudiados 77 pacientes con estudios básicos de hemostasia, pruebas específicas alteradas, y coagulopatías diversas. El 55% (43) fueron al sexo masculino.
La mediana de edad fue de 18 años, (rango de 1 a 75) años. Al estratificar por categoría de edad, el 53% de los pacientes eran menores de 18 años (Tabla 3).
Tabla 3 Frecuencia de edades por grupos etarios
| Edades | Frecuencia | Porcentaje |
| De 1 - 18 años | 41 | 53 |
| De 19 - 75 años | 36 | 47 |
| Total | 77 | 100 |
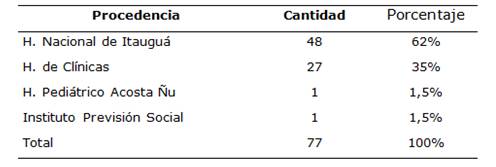
En cuanto a la procedencia hospitalaria de los pacientes, la mayoría fueron del Hospital Nacional (62%) (Tabla 4).
El 90% (69) de los pacientes fueron ambulatorios y 5% (8) internados.
Deficiencias de la vía extrínseca de la coagulación
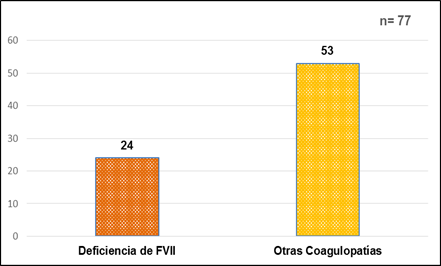
El 31% (24/77) de los pacientes estudiados, mostraron una deficiencia del factor VII (FVII) de la coagulación (Fig. 1).
El 100% de los resultados de la Deficiencia de FVII (24/24), fueron leves de valores entre 12 y 59% para un valor de referencia de 70 a 120% y un nivel hemostático de 10 a 20 %.
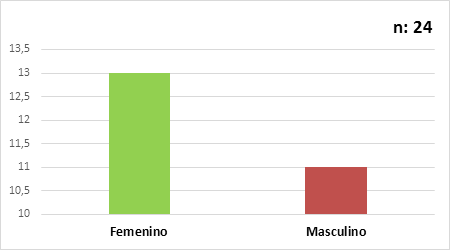
La edad del diagnóstico en estos pacientes con deficiencia de factor VII, se encontraba entre 7 y 75 años con una mediana de 18 años, ligero predominio del sexo femenino, F: 13/24 y M: 11/24 (Fig. 2).
Deficiencias de la vía intrínseca de la coagulación
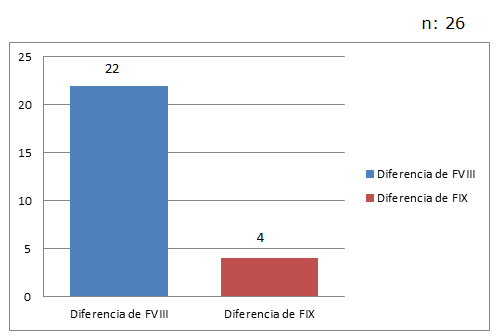
La deficiencia de Factor VIII o Hemofilia A, fue la más observada con un 29% (22/77), mientras que la deficiencia del factor IX o Hemofilia B se observó en 4 pacientes, con un 5% (4/77)
De los 26 pacientes con Hemofilia, se observó 85% (22/26) de Hemofilia A y 15% (4/26) de Hemofilia B. (Fig. 3).
Las edades de los pacientes con ambas deficiencias variaron entre 1 y 64 años con una mediana general de 13,5 años.
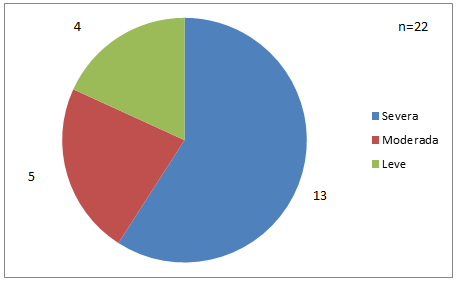
Se encontraron 13 pacientes con Hemofilia A severa, 5 con Hemofilia A moderada y 4 con Hemofilia A leve (Fig. 4).
Cuatro pacientes fueron diagnosticados con deficiencia de factor IX o Hemofilia B, y en cuanto a gravedad en el momento del diagnóstico, se observaron 3 pacientes con FIX entre 1 y 5% y un paciente con FIX inferior a 1%. Las edades de estos pacientes variaron entre 4 y 54 años.
No se observaron deficiencias de factor XI y XII, en los pacientes estudiados, durante el periodo de estudio.
Deficiencias de la vía común final
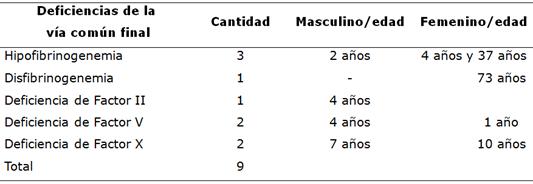
Se han observado deficiencias de la vía común en 9 pacientes: 4 han tenido deficiencias del Factor I o Fibrinógeno; 3 con hipofibrinogenemias y 1 con probable disfibrinogenemia, 1 paciente con deficiencia del Factor II, 2 pacientes con deficiencias del Factor V, y 2 pacientes con deficiencias del Factor X de la coagulación (Tabla 5).
Respecto a sospecha de inhibidores
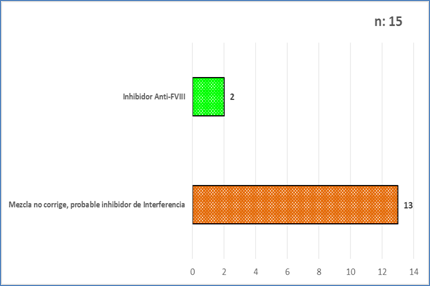
Se observaron 15 resultados de pacientes con sospecha de inhibidores, se confirmaron 2 pacientes con inhibidor específico Anti-FVIII, la titulación por el método de Bethesda, dio como resultado 26 UB/mL, paciente masculino, 29 años y el otro 160 Unidades, paciente masculino de 15 años; para un valor de referencia de <0,6 UB/mL; ambos pacientes eran portadores de Hemofilia A severa. Los otros 13 pacientes, con sospecha de inhibidor de interferencia, orientado por la mezcla con plasma normal que no corrigió y que, en las pruebas de dilución fue aumentando la actividad del factor a medida que aumentaba la dilución de la muestra, característica de los inhibidores de interferencia (Fig. 5).
Enfermedad de Von Willebrand
Se tuvieron 4 pacientes con sospecha de Enfermedad de Von Willebrand. Al momento de esta investigación se estaba poniendo a punto la prueba, y se confirmó esta deficiencia, en una paciente de sexo femenino, de 47 años, con un valor de VWF Ag. 11% y VWF Act. 12%, FVIII 16,5%, con antecedentes de sangrado.
DISCUSIÓN
De 77 pacientes estudiados, 43 fueron del sexo masculino, esto pudo ser debido a la cantidad de las deficiencias del factor VIII (Hemofilia A) y factor IX (Hemofilia B) halladas, los cuales se heredan con rasgo recesivo ligado al sexo, en 1 a 3 varones cada 10.000 y 1 cada 30.000 varones respectivamente 12,13.
Los pacientes provenían del Hospital Nacional de Itauguá y del Hospital de Clínicas, instituciones que cuentan con médicos hematólogos adultos y pediátricos, quienes realizaron los pedidos orientados hacia probables coagulopatías.
En la presente investigación, el 90 % de los pacientes fueron ambulatorios, lo cual era de esperar, ya las pruebas básicas del coagulograma se solicitan de rutina, como un análisis de control prequirúrgico, o la repetición ante el resultado de un tiempo de protrombina (TP) bajo, a través de un tamizaje del sistema de coagulación6.
Se registró 31% de deficiencias de factor VII en la población estudiada, todos ellos de severidad leve; pero se ha visto que no hay relación entre la concentración del factor y la manifestación clínica10,14. Por lo que estas cifras constituirían un sub registro, ya que probablemente existan muchas personas con estas deficiencias, quienes al no tener manifestación clínica no consultan.
La deficiencia de factor VII es la más frecuente de los trastornos de la coagulación poco comunes, la deficiencia grave ocurre en 1 persona en 500.000 entre la población en general10,11,13,15. Entre los pacientes con déficit de FVII se vió un ligero predominio del sexo femenino, F: 13/24 y M: 11/24. Las hemorragias en membranas mucosas, incluyendo epistaxis y menorragia, son comunes10. Razones por las que a veces y sobre todo las mujeres consultan y son derivadas al médico hematólogo para su investigación.
En cuanto a las deficiencias de la vía intrínseca, el FVIII o Hemofilia A, fue la más observada 29% (22/77), y la deficiencia del FIX o Hemofilia B, 4 pacientes, 5% (4/77). La edad varió de 1 a 64 años, mediana de 13,5 años. Para los casos graves, se debería diagnosticar por lo menos al año de vida, cuando existe mayor movilidad y se evidencia por hemorragias en sitios de presión y apoyo, para los casos moderados y leves el diagnóstico se retrasa a veces hasta la edad adulta, al presentarse hemorragia posterior a una agresión externa13.
Todos los pacientes diagnosticados con hemofilia fueron del sexo masculino; debido a que las deficiencias de estos factores son heredadas de forma recesiva, ligado al sexo, los hombres son los afectados, mientras que las mujeres son las que portan y transmiten la enfermedad14,16,17.
Según la Federación Mundial de Hemofilia (FMH), la prevalencia de la hemofilia A informada en países de ingresos bajos es a menudo considerablemente menor que la informada en países de ingresos elevados, y menor, a la esperada con base en la incidencia internacional promedio18.
Nuestro país no tiene un registro oficial de pacientes con Hemofilia a nivel estatal, actualmente existe un indicio de trabajo a través del Programa Nacional de Sangre y la Fundación de ayuda al Hemofílico (Funda Hemo)19 dentro del cual el Hospital Nacional de Itauguá, fue escogido como Centro de referencia para el diagnóstico y tratamiento de pacientes en situación de Hemofilia, a partir del cual se espera obtener un registro de dichos pacientes.
En el estudio se halló 85% (22/26) de Hemofilia A y 15% (4/26) de Hemofilia B. Según Martínez-Murillo( 16 ), la Hemofilia A se presenta en el 80% de todos los casos, contra el 20 al 25% de casos de Hemofilia B.
Según la FMH algunas razones posibles para el informe de un menor número de casos de hemofilia podrían ser: que la mayoría de los hemofílicos del mundo: 1. no han sido identificados debido a una falta de capacidad diagnóstica, 2. no tienen acceso a la atención, 3. no tienen recursos económicos, y 4. tienen poca o nula disponibilidad de terapia de reemplazo de FVIII. Sin tratamiento, las personas que padecen hemofilia severa con frecuencia mueren durante la infancia o la vida adulta temprana, lo que da por resultado una menor prevalencia en relación con el número de casos nacidos18.
No se observaron deficiencias de factor XI y XII, en los pacientes estudiados, durante el periodo de estudio. Estas deficiencias también son poco frecuentes, la deficiencia del factor XI se manifiesta por prolongación del TTPA, y a veces solo las deficiencias graves presentan una leve tendencia a la hemorragia. Mientras que la deficiencia de factor XII (FXII) no genera un trastorno de la coagulación, la deficiencia (heterocigoto) es común entre la población caucásica en general (2,3% de los donantes de sangre) y es la causa más común de un TTPA prolongado inesperado durante las pruebas prequirúrgicas10,12,13.
De la Vía común, las deficiencias encontradas fueron: cuatro pacientes con alteraciones del fibrinógeno o FI, 3 hipofibrinogenemias y 1 probable disfibrinogenemia, 1 paciente con déficit de FII, 2 de FV y 2 de FX.
Las anormalidades del fibrinógeno pueden ser ausencia de fibrinógeno, afibrinogenemia, nivel reducido de fibrinógeno, con estructura normal: hipofibrinogenemia, fibrinógeno estructuralmente anormal: disfibrinogenemia. En la práctica puede ser difícil distinguir entre la hipo y la disfibrinogenemia. Las formas leves probablemente son subdiagnosticadas. Los trastornos del fibrinógeno con manifestaciones hemorrágicas graves son poco comunes10.
El déficit del factor II es infrecuente, se transmite de forma autosómica recesiva10,12,13,20. La deficiencia completa del FII parece ser incompatible con la vida. En la mayoría de los casos se trata de deficiencias tipo I (disminución del FII antigénico y funcional), aunque hay descritos casos de disprotrombinemia. Se caracteriza por sangrado después de maniobras invasivas (incluyendo la caída del cordón umbilical), hemartrosis, hematomas musculares y sangrado mucoso13.
El déficit congénito de FV es una enfermedad rara que se transmite de manera autosómica recesiva, las formas homocigotas varían desde sangrados menores, a formas que sólo se manifiestan en caso de cirugía10,12,13,20.
La deficiencia grave de FX (FX <1 UI/dl) es por lo general un trastorno de la coagulación grave; sigue una herencia autosómica recesiva. También es poco frecuente en su forma grave, afectando a 1/1.000.000 personas, conlleva un riesgo particular de hemorragia intracraneana o HIC durante el periodo neonatal10,12,13,20.
La deficiencia de los factores de la vía común es poco frecuente, los factores; FI, FV y FX de 1/1.000.000 de personas de la población general, y FII de 1/2.000.000.
Se registraron 15 pacientes con probables inhibidores de los cuales, 2 se confirmaron como Anti-factor VIII, ambos hemofílicos tipo A severos. Cerca del 30% de los hemofílicos que inician un tratamiento con factor VIII van a desarrollar un inhibidor contra esta proteína de la coagulación, lo que va a dificultar su tratamiento. La proporción es claramente más baja en los hemofílicos B, entre los cuales solamente el 5% desarrollará esta complicación18,20,21.Con respecto a la frecuencia de inhibidores es variable, en el caso de la hemofilia A varía entre 3 y 52%, aunque se considera que entre 20 y 30% de los pacientes con hemofilia severa desarrollará un inhibidor y será menos frecuente en los tipos leve-moderado18,22-25.
Los restantes 13 pacientes con probable inhibidor de interferencia; que podrían ser el inhibidor lúpico que es la más frecuente26. Se debería realizar otros estudios confirmatorios para inhibidores de interferencia; en la sección de hemostasia se puede llegar hasta la sospecha.
Se registraron 4 pacientes con sospecha de Enfermedad de Von Willebrand, confirmándose en una paciente de sexo femenino 47 años. Es un trastorno congénito transmitido autosómicamente, caracterizado por el déficit cualitativo y/o cuantitativo del factor de von Willebrand (FvW). Es la diátesis hemorrágica más frecuente, habiéndose calculado que en determinadas áreas puede llegar a afectar al 1% de la población, aunque los pacientes con problemas hemorrágicos graves no son numerosos13. Se deben realizar numerosas pruebas para el diagnóstico de la enfermedad de von Willebrand, como las pruebas básicas o globales, que serán normales excepto en combinaciones con otros defectos y las pruebas específicas que investigan el FvW (funcional e inmunológico) y el FVIII, necesarias para confirmar el diagnóstico27-30.
Estos pacientes fueron derivados de médicos hematólogos con otras pruebas generales normales, y clínica hematológica compatible con la enfermedad de von Willebrand. En nuestro país no existen registros sobre la enfermedad de von Willebrand, por lo que se considera resaltante este hallazgo.
CONCLUSIÓN
Se reclutaron 77 pacientes con coagulopatías en el Departamento de Laboratorio del Hospital Nacional de Itauguá, ligero predominio masculino, de 1 a 75 años, mediana de 18 años.
Entre las coagulopatías hemorrágicas de mayor frecuencia, están en 1er lugar las Hemofilias A y B, seguida de la deficiencia del Factor VII, y luego los probables inhibidores de interferencia, los dos casos de inhibidores específicos anti Factor VIII fueron en pacientes con Hemofilia A severa.
Fue relevante el hallazgo de deficiencias de factores dela vía común de la coagulación, a pesar de ser poco frecuentes.
Se destaca que muchas deficiencias de factores se realizaron en la edad adulta de los pacientes estudiados, reflejando lo tardío que se llega al diagnóstico en nuestro país.

