










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista del Nacional (Itauguá)
Print version ISSN 2072-8174
Rev. Nac. (Itauguá) vol.7 no.1 Itauguá June 2015
https://doi.org/10.18004/rdn2015.0007.01.046-049
CASO CLINICO
Síndrome prune belly (vientre en ciruela): reporte de un caso
Prune belly syndrome(belly plum): a case report
Andrea Carolina Portillo1
RESUMEN
Se presenta caso clínico de una mujer gestante quien en su primera consulta prenatal, con una gestación pretérmino por examen físico, presenta una ecografía que informa malformaciones en el sistema urinario y digestivo fetal. En el Hospital Nacional (Itauguá) se confirman dichos hallazgos. El desenlace fue fatal al mes de nacido, por complicaciones en la cirugía intervencionista intestinal.
Palabras claves: síndrome prune belly, pseudo prune belly, malformación congénita
ABSTRACT
Clinical case of a pregnant woman who in her first prenatal visit, with a preterm gestation by physical examination, fetal malformations in the urinary and digestive system is presented in the ultrasound. In the National Hospital of Itaugua these findings are confirmed. The outcome was fatal a month after the pacient was born, due to complications in the intestinal interventional surgery.
Key words: prune belly syndrome, pseudo prune belly, congenital malformation
INTRODUCCION
El Síndrome de prune belly ó abdomen en ciruela ó triada de Eagle Barret (1.950) es un complejo sintomático, descripto por primera vez en el año 1.839 por Forhlich1-3. Su incidencia es 1/35.000-50.000 nacidos vivos4-6, con mayor prevalencia en el sexo masculino (18-20/1) ó 97% vs 3-4% en relación al sexo masculino y femenino7. La mayoría de los casos se encontró en madres menores a 30 años.
La triada clásica es la distensión abdominal, por hipoplasia ó atrofia muscular, anormalidades del tracto urinario del tipo obstructivo (megavejiga, megaureter, hidronefrosis de grado variable, displasia renal de grado variable) y criptorquidia bilateral8-12.
A la triada característica se ha asociado otras malformaciones: en el sistema musculoesquelético (50 %), sobre todo en los casos cuando hay oligoamnios, aparecen pie talipers, pie equino varo, pie zambo, displasia de cadera, escoliosis, artrogriposis, tortícolis, etc. Las cardiopatías (10%) son: defecto septal auricular, defecto septal ventricular, ductus arterioso permeable, tetralogía de Fallot. En aparato respiratorio hay hipoplasia pulmonar, resultante del compromiso relacionado al aumento del volumen torácico a expensas del oligoamnios y la distensión abdominal. En tubo digestivo (30%) se detecta: malrotación intestinal, atresia ó estenosis intestinal, ano imperforado, cloaca persistente, gastrosquisis, onfalocele, vólvulos, enfermedad de Hirschsprung. En sistema nervioso central hay ventriculomegalia. Otras malformaciones son: fascie de Potter, micrognatia, hipertelorismo, fisura palpebral, puente nasal aplanado, orejas de implantación baja, éstos últimos asociados al oligoamnios severo1.
La etiopatogenia, aunque incierta aún, se han atribuido a 3 teorías8,9,11:
1. Por una noxa (teratógenos, drogas como la cocaína, incluso ser multifactoria) que interrumpe el normal desarrollo del mesodermo: placa intermedio lateral entre la 6º a 10º semana de gestación. Esta teoría no explica la criptorquidia ni anomalías ureterales. Por lo que la más aceptada es la siguiente:
2. Por un fenómeno obstructivo, malformación uretral en cierto periodo de formación, donde la uretra anterior y posterior2,3 por procesos obstructivos crea un sistema de alta presión en todo el tracto urinario, dando uréteres dilatados y tortuosos, vejiga agrandada, hidronefrosis de grado variable y, secundariamente, una laxitud de la pared abdominal7.
3. Una teoría reciente es la persistencia del saco vitelino/alantoides y la sustitución del mesénquima por somitas dorsales ó lumbares.
En general, la causa es aún controvertida. Actualmente se atribuye que la causa es genética (mutación homocigota del cromosoma 1 q43)12 pues se ha documentado casos familiares. Pero la mayoría de los casos de Pseudo prune belly son esporádicos, no familiares7.
El pronóstico principal está dado por el grado de compromiso del tracto urinario8,9. El defecto de la pared abdominal por sí mismo no tiene significado pronóstico11.
El manejo prenatal está destinado al estudio del cariotipo para evaluar su recurrencia9, mejorar la función renal, previa selección de candidatos para su intervención, con la introducción de un catéter doble pigtail entre la vejiga y la cavidad amniótica4,6,10. En los infantes, se realiza el tratamiento médico de las complicaciones como infecciones, a veces puede requerir intervención quirúrgica de diversas modalidades, desde vesicostomía temprana hasta trasplante renal en los casos de fallo renal severo. El abordaje quirúrgico específico incluye abdominoplastia reconstructiva, orquidopexia bilateral, cistoplastia reductora y reimplantación ureteral9.
Presentación de un caso
Paciente de 20 años de edad, secundípara, gestante de pretérmino por examen físico, fecha última menstruación dudosa, acude a su primer control prenatal, donde le solicitan ecografía. Es resultado informa feto vivo, estimado en 35 semanas por ecobiometría, con hallazgos malformativos: vejiga dilatada, hidronefrosis y polihidramnios.
Padres no consanguíneos, antecedente de 1 aborto espontáneo hace 4 años. Se solicitan análisis y remite al Hospital Nacional de Itauguá.
Durante su instancia en el Hospital Nacional se recibe resultados de STORCH / HIV arrojando resultados negativos; hemoglobina 13 mg/dL.
Se repite ecografía en el Servicio de Obstetricia (fig. 1, 2 y 3) informando megavejiga, megaureter, hidronefrosis e imagen quística por arriba de la vejiga que podría corresponder a intestino ó estómago.
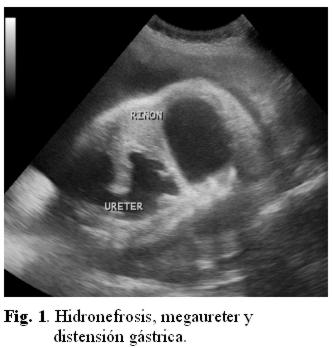
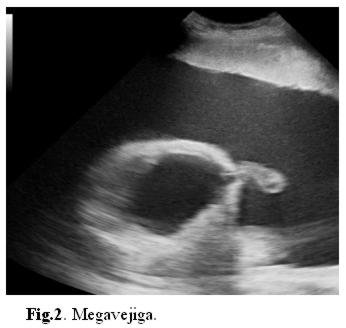
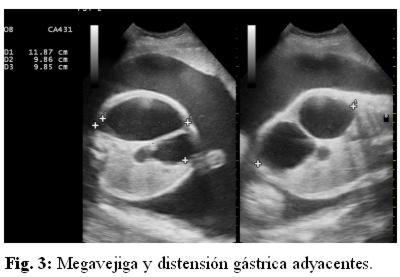
Se decide interrupción de la gestación por sospecha de alteración del bienestar fetal por cesárea. Se da a luz un recién nacido de sexo femenino, Apgar 5/7 a expensas de reflejo, tono, respiración y color, peso 2.500 gr, de 36 semanas por Capurro. Necesita estimulación y oxígeno al nacer. Se aspira líquido meconial fluido en abundante cantidad. El examen físico neonatal reveló (fig. 4): abdomen asimétrico a expensas de tumoración en epigastrio, diastasis de músculos rectos anteriores, resto del examen físico: normal.
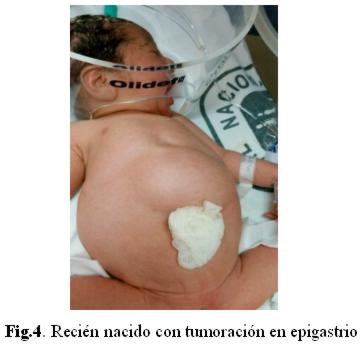
El paciente fue trasladado a Unidad de cuidados intensivos para estabilización e inicio de estudios, con alimentación enteral y sonda vesical en permanencia.
Se le realiza ecografía abdominal (fig.5) que informa ambos riñones aumentados de tamaño con dilatación pielocalicial, vejiga muy distendida, hígado, bazo y vesícula normales.
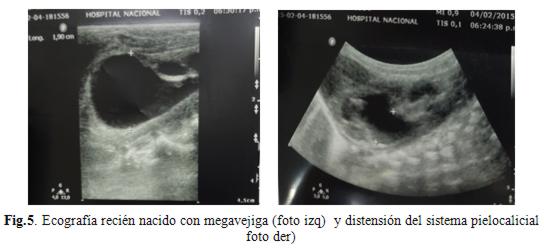
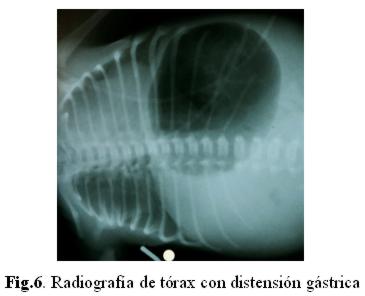
La ecografía transfontanelar es normal. Su perfil renal es normal. Se realiza placa de abdomen (fig.6) sonde se evidencia estómago muy distendido, sin pasaje de aire a través del intestino.
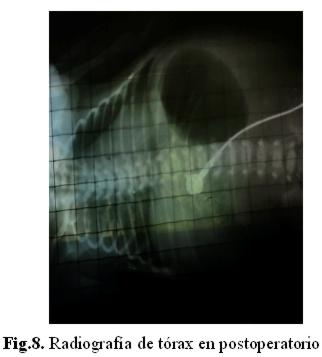
En su 2º día de internación, en interconsulta con cirujanos pediátricos, se decide laparotomía exploradora, hallándose atresia yeyunal distal. Se realiza yeyunostomía término-lateral según técnica de Santulli, se halló intestino proximal normal y vejiga muy distendida. El recién nacido (fig. 7 y 8) permanece hemodinámicamente estable con asistencia mecánica respiratoria, y decide administración profiláctica de ampicilina + gentamicina.
En su 7º día del postoperatorio aparece distensión abdominal e ileostomía no funcionante, por lo que los cirujanos realizan estimulación de la ostomía con suero fisiológico; se rota antibiótico a una cefalosporina de 3º generación debido a leucocitosis.
Al 9º día del postoperatorio se constata abdomen asimétrico a expensas de distensión gástrica, más ileostomía no funcionante, por lo que deciden re-laparotomía. El hallazgo quirúrgico es una membrana pre-pilórica más estenosis de la anastomosis ileal; se realiza cirugía de Mickher más reconstrucción ileal con ileostomía proximal.
El líquido cefalorraquídeo fue normal, en hemocultivos se aisló Burkholderia cepacia. Paciente con aparente buena evolución excepto por radiografía de tórax que revela una atelectasia unilateral, que luego recupera.
A los 22 días post-laparotomía, el paciente se presenta polipneico, con alto débito por sonda nasogástrica, abdomen distendido, doloroso, ruidos hidroaereos disminuidos, ostomía no funcionante por lo que deciden nuevamente intervención quirúrgica, cuyo hallazgo fue dehiscencia de la anastomosis termino-lateral, peritonitis fecal y fístula ileodistal.
Paciente presenta una evolución tórpida, aumento de glóbulos blancos, anemia y alteración del coagulograma. Se realiza diagnóstico de sepsis a punto de partida intestinal. A pesar de rotar antibiótico a piperacilina, corregirse la anemia y plaquetopenia, la paciente no mejora, se halla deshidratada, edematosa, con parámetros altos en la asistencia respiratoria mecánica, con abdomen distendido y doloroso. A los 34 días de nacida sigue con cuadro de acidosis metabólica descompensada, presenta paro cardiaco, pero a pesar de las maniobras de reanimación, fallece.
COMENTARIO
El Síndrome de prune belly es una rara anomalía congénita que puede ser sospechado in útero por medio de la ecografía. El recién nacido no manifestó compromiso en la función renal a pesar del compromiso obstructivo, como la mayoría de los casos publicados. Hay que tener en cuenta las malformaciones asociadas, que muchas veces son factores sumatorios en el destino del paciente como representó el caso presentado, a través de complicaciones en la intervención quirúrgica intestinal. Posiblemente falleció por sepsis de origen intestinal.
REFERENCIAS
1. Gallo M, Sánchez R, Gallo J, Ruoti Cosp M, Hernández A. Ecografía fetal de semana 18-22 de embrarazo: colección de medicina fetal y perinatal. 1º ed. Venezuela: Amolca. 2014. cap. 8. p. 155. [ Links ]
2. Gallo M, Martínez-Ten P, Espinoza A. Ecografía tridimensional (3D-4D) en embarazo.1ª ed. Venezuela: Amolca. 2013. cap. 17. p. 367-368. [ Links ]
3. Mahuad Filho F. Cunha Ferreira A. Naves do Amaral W. Ultrasonografía en ecografía y obstetricia: guía práctica. 1ª ed. Venezuela: Amolca. 2012. cap. 32. p 168-169. [ Links ]
4. Pastore A. Ultrasonografía en ginecología y obstetricia: t 1. Venezuela: Amolca. 2012. cap.28. p 380-381. [ Links ]
5. Avni FE, Maugey-Laulom B, Cassart M, Eurin D. et al. Aparato genitourinario fetal. En: Callen P. Ecografía en obstetricia y ginecología. 5ª ed. Barcelona: Elsevier Masson, 2009. p. 640-675. [ Links ]
6. Gratacós E. Gómez R. Nicolaides K. Romero R. Cabero L. Medicina Fetal. 1ª ed. Argentina: Panamericana. 2009. p. 449-450. [ Links ]
7. Hoyos Orrego A, Botero González P. Síndrome de prune belly (ciruela pasa): reporte de un caso. Medicina UPB 2010;29(2):155-161 [ Links ]
8. Mata-García LE, Chávez-Ocaña S. Síndrome de prune belly: revisión de literatura a propósito de un caso. Rev Hosp Jua Mex. 2013;80(2):134-137. [ Links ]
9. Logroño Di Vianna R, Montero Ramos JP. Síndrome de prune belly (vientre en ciruela). Reporte de caso y revisión de la literatura. Acta Medica Dominicana. 1993;15(4):130-135. [ Links ]
10. Maita Quispe F, Panozo Borda SV, Verástegui Céspedes DE, Hochstatter Arduz EA, de Guzmán ON, Zegarra Santiesteban W. Síndrome de Prune Belly: diagnóstico y manejo pre y posnatal, presentación de dos casos. Gac Med Bol 2013;36(1):35-38. [ Links ]
11. Durán Padilla MA, Rivero de Jesús V, Macías Jiménez B. La variante letal del síndrome Prune Belly. Informe de dos casos. Rev Med Hosp Gen Mex. 1999; 62(3): 206-209. [ Links ]
12. Tattoli F, De Prisco O, Gherzi M, Falconi D, Marazzi F, Marengo M, et al. Prune-Belly Syndrome: a case report. G Ital Nefrol. 2014 May-Jun; 31(3). pii: gin/31.3.5. [ Links ]
1. Gineco-obstetra. Departamento de Gineco-obstetricia. Hospital Nacional. Ministerio de Salud Pública y Bienestar Social (Itauguá - Paraguay)
Correo electrónico: andreaporti93@gmail.com
Artículo recibido: 20 de abril de 2015. Artículo aprobado: 17 de mayo de 2015.
