










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista del Nacional (Itauguá)
Print version ISSN 2072-8174
Rev. Nac. (Itauguá) vol.4 no.1 Itauguá June 2012
CASO CLINICO
Síndrome de activación macrofágica idiopática en una mujer jóven
Idiopathic macrophage activation syndrome in a young woman+
*Luis Benítez Luis1, Dora Montiel-Jarolín1, Estela Torres de Taboada1, Luz Negri1 Nidia Aquino1,Ángeles Barrios1
1. Departamento de Medicina Interna. Hospital Nacional (Itauguá, Paraguay)
Artículo recibido: 13 de Abril de 2012. Aprobado: 19 de Mayo de 2012
RESUMEN
Se presenta el caso de una mujer de 27 años, con datos clínicos de síndrome de activación macrofágica (SAM), de etiología desconocida, con evolución tórpida y desfavorable que llega al fallecimiento. Se revisan los principales conceptos y características del SAM, haciendo énfasis en las actuales normas de consenso y en las variaciones en el tratamiento de acuerdo con las guías internacionales.
Palabras clave: hemofagocitosis, médula ósea, corticosteroides, fiebre prolongada, citopenia.
ABSTRACT
We report the case of a 27 year old woman with clinical evidence of macrophage activation syndrome (MAS) of unknown etiology and unfavorable torpid comes to death. We review the main concepts and features of the SAM, the current emphasis on consensus standards and changes in treatment according to international guidelines.
Keywords: hemophagocytosis, bone marrow, corticosteroids, prolonged fever, cytopenia.
INTRODUCCION
El síndrome de activación macrofágica (SAM) es una enfermedad infrecuente, de evolución fatal, secundaria a la activación del sistema fagocítico mononuclear, que se caracteriza por la proliferación incontrolada de los histiocitos con fenómeno de hemofagocitosis. Es un desorden de la regulación inmune caracterizado por fiebre, hemofagocitosis, hepatoesplenomegalia, pancitopenia, hipertrigliceridemia y coagulopatía. La etiología del SAM es desconocida pero se describen factores desencadenantes tales como infecciones virales y drogas. Actualmente, el tratamiento de elección son los corticoesteroides endovenosos.Objetivo: presentar una paciente con síndrome de activación macrofágica idiopática internada en el Hospital Nacional de Itauguá. Caso Clínico: mujer de 27 años, de profesión secretaria, procedente de Capiatá, dos hijos, divorciada, sin antecedentes patológicos de interés.Consulta en agosto 2011 por cuadro de 6 meses de evolución que inicia con sensación febril, no graduada, de inicio insidioso, intermitente, de predominio nocturno, que cede con antipiréticos comunes (paracetamol, dipirona). Refiere además pérdida de peso, de 3-4 Kg. aproximadamente y debilidad generalizada desde el inicio del cuadro. Ante la persistencia de los síntomas, consulta con facultativos, quienes indicaron estudios, donde se constató anemia y es tratada con antipiréticos sin mejoría del cuadro. Niega artralgias, mialgias, caída de cabello, ingesta de fármacos, contacto con animales y antecedente de viajes. No se conoce alérgica, asmática, hipertensa ni diabética. Refiere cirugía anterior: amigdalectomía hace 1 año por faringoamigdalitis a repetición. Consumidora de productos naturales Herbalife©, con el objetivo de bajar de peso, desde hace 1 año. No fuma ni bebe alcohol, no es consumidora de drogas ilícitas.
Examen Físico: temperatura 38°C, palidez de piel y mucosas, impresiona punta de bazo a la palpación abdominal y lesiones nodulares a nivel del tejido celular subcutáneo interpretándose como lipomas, resto del examen físico sin datos de interés.
Laboratorio: Hemograma: glóbulos blancos: 2600/ mm3, neutrófilos 76%, linfocitos 8%, monocitos 15%, hemoglobina 9,6 g/dlL, hematocrito 28%, volumen corpuscular medio 85 µm3, plaquetas 200.000/mm3, eritrosedimentación 45 mm. Glucosa 94mg/dL, urea y creatinina normales. Proteina C reactiva 5,6 mg/dL, VDRL negativo, lactato deshidrogenasa 350 UI/L, hierro sérico 43 mcg/dL, transferrina 227 mg/dL, proteínas totales 7 g/dL, albúmina 2,2 g/dL, aspartato aminotransferasa 94 UI/L, alanina aminotransferasa 35 UI/L, fosfatasa alcalina 64 U/L, hepatograma normal. Crasis sanguínea normal. Orina simple no patológica. Hemocultivos: 3 muestras negativos, urocultivo negativo. Serologías: HIV negativo, Hepatitis A-B-C: negativos, Citomegalovirus IgM negativo e IgG 27 UI/mL. Herpes IgG e IgM negativos, Chagas IgM e IgG negativos, Factor reumatoideo negativo, Anti DNA negativo. C3: 2,4 mg/dL. Antígeno RK39 negativo. Antígenos febriles negativos. Anticuerpo antiestreptolisina negativo. ANCA-p negativo, ANCA-c negativo. Anticoagulante lúpico negativo, T3, T4 y TSH normales. Crioglobulinemia negativo. Cultivo de ganglio axilar negativo. Cultivo de quiste de Bartolino negativo.
Radiografía de Tórax: normal (al ingreso). Ecografía Abdominal: dentro de parámetros normales. No se observan visceromegalias. Evolución: Permanece internada en nuestro hospital por 1 semana. Durante su internación se presentó febril y posteriormente pide alta solicitada para ser estudiada en otro centro asistencial privado, donde es internada el mismo día de su salida de nuestro hospital. La paciente permanece en el centro asistencial privado por 14 días, donde continuó febril. Se realizó una punción aspiración de médula ósea que informó: medula ósea celular, de carácter reactivo, con detritus (+) en macrófagos.Tres días después de su ingreso presentó tos seca y dificultad respiratoria progresiva. El cuadro respiratorio se interpretó como una neumonía. Es tratada con ceftriaxona-ciprofloxacina (que recibió por 1 semana). Debido a la evolución desfavorable de la paciente, se decidió su traslado a Unidad de Terapia Intensiva del Hospital Nacional (02/09/11). La paciente ingresó lúcida, febril, con dificultad respiratoria a mínimos esfuerzos, hemodinámicamente inestable. Se decidió rotar de antibiótico a cefoperazona-sulbactam cubriendo una neumonía intrahospitalaria. Permaneció en Terapia 24hs, sin asistencia respiratoria mecánica, y luego es trasladada Clínica Médica.
03/09/12: La paciente presentó leve mejoría de la disnea, se mantuvo febril en forma persistente, con hemodinamia estable. Nuevos datos de laboratorio: frotis de sangre periférica policromasia (+), poiquilocitosis (+), hipocromia (++), anisocitosis (+). Orina simple: normal. Test de Coombs directo negativo. Electroforesis de las proteínas: alfa 1 globulina: 16,4% g/dL (VN 2,9-4,9), alfa 2 globulina 18,4 g/dL (VN 7,1-11,8), gammaglobulinas: hipergamma policlonal. Crasis sanguínea: tiempo de protrombina: 17,1 seg, tiempo de protrombina (actividad): 56%, tiempo parcial de tromboplastina activada 32 seg, fibrinógeno 57 mg/dL, tiempo de trombina 41 seg.
23/09/11: La paciente presentó dificultad respiratoria a mínimos esfuerzos, con cianosis labial y distal. Se agregó al tratamiento antibiótico para ampliar espectro: ciprofloxacina-ceftriaxona. Al examen físico: Pulmones: crepitantes bilaterales hasta campo medio. Piel: rash cutáneo pruriginoso a nivel de tronco y miembros. Debido a la aparición de lesiones en piel, se decidió suspender ceftriaxona y se agregó vancomicina al tratamiento, por sospecha de efectos colaterales (rash cutáneo). Paciente presentó mejoría clínica, permaneciendo apirética por 3 días, sin disnea, pero persistiendo el rash cutáneo.
27/09/11:El cuadro febril reapareció, llegando a graduarse la temperatura en 41°C y las lesiones a nivel de piel se exacerbaron, tornándose purpúricas y generalizándose, respetando el rostro (figura 1). Se realizó el diagnóstico de síndrome purpúrico de etiología a determinar.

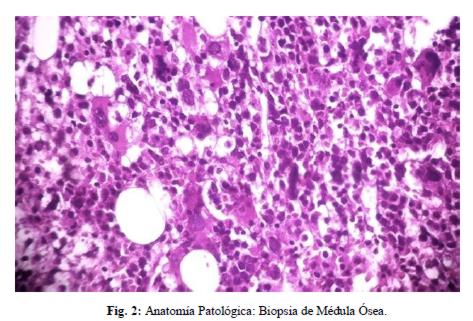
La paciente presentó inestabilidad hemodinámica y mala mecánica respiratoria, se constató descenso de plaquetas, empeoramiento de las lesiones purpúricas, por lo que recibió trasfusión de plaquetas y se decidió su traslado a sala de reanimación. El laboratorio informa plaquetas: 102.000/mm3 (22/09), luego 80.000/mm3 (26/09) y 46.000 /mm3 (27/09). En reanimación, la paciente respondió a hidratación generosa y se inició trasfusión de glóbulos rojos, plasma y plaquetas. No se consideró cambio de cobertura antibiótica. Se realizó biopsia de medula ósea que informó: médula hipercelular, hallazgos compatibles con displasia medular, con inmunohistoquímica negativa para linfoma. (figura 2)
29/09/11:La paciente se trasladó a sala de Clínica Médica, persistiendo febril, con dificultad respiratoria, se tomaron cultivos y se asoció al tratamiento ceftazidima. Cuatro hemocultivos y un urocultivo retornan negativos. Recibió pulsos de metil prednisolona 1gr/día por 3 días, seguidos de 100 mg/día de prednisona.
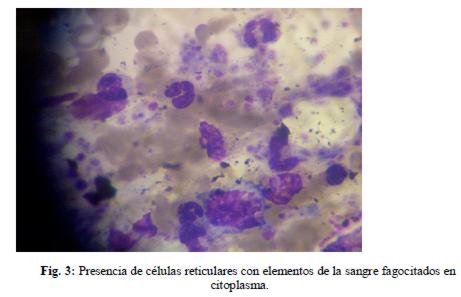
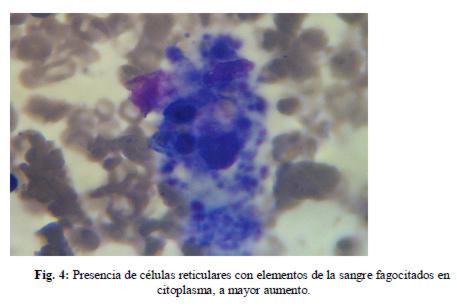
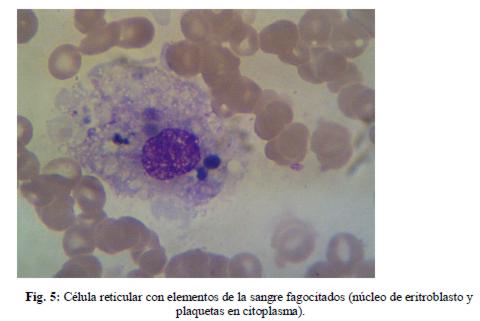
10/10/11: La paciente evolucionó febril, por lo que se realizó nueva punción medular y posteriormente se trasladó a Terapia, recibiendo perfusión de inmunoglobulinas. Se recibió luego resultado de mielograma que informó: médula celular, cambios displásicos, activación de sistema reticular con hemofagocitosis.(figuras 3, 4 y 5)
13/04/12: La paciente evolucionó tórpidamente en terapia presentando falla multiorgánica refractaria al tratamiento y fallece.
El síndrome de activación macrofágica no fue sospechado en nuestra paciente, a pesar de reunir todos los criterios diagnósticos.Fue tratada con pulsos de metil prednisolona y perfusión de inmunoglobulinas con mala evolución.
DISCUSION
En la presente comunicación hemos dado a conocer un caso de una paciente que ha presentado síndrome de activación macrofágica (SAM) o síndrome hemofagocítico secundario (SHFS). El SAM corresponde a un conjunto de síntomas clínicos causados por una excesiva activación y proliferación de macrófagos bien diferenciados que fagocitan elementos hematopoyéticos y diferentes linajes celulares maduros, asociado a una intensa proliferación de linfocitos T con la consecuente liberación de citoquinas. Este síndrome es extraordinariamente raro en la práctica y su primera descripción data del año 1985 cuando fue publicado el primer caso en un paciente portador de una Artritis Idiopática Juvenil de inicio sistémico. El término de SAM, sin embargo, en el campo de la hemato-oncología, una condición clínicamente similar se define como "linfohistiocitosis hemofagocítica reactiva" (LHHR). Existen diferentes condiciones que llevan a que este proceso se desencadene, entre los que se distingue un proceso de alteración genética y varias formas reactivas, a entidades de alteración del sistema inmunológico, infecciones o a enfermedades malignas.
Entre las divisiones de las formas primarias y secundarias podemos destacar: (ver tabla 1).

Los criterios diagnósticos son5,6 (Tabla 2)

Las normas terapéuticas se basan en las guías de los estudios Hemophagocytic Linpho-Histiocytosis (HLH) de 1994 y de 2004, que recomiendan las siguientes medidas terapéuticas: 6
Estudio HLH-1994:7
Inicio de tratamiento con dexametasona y etopósido.
Tratamiento con ciclosporina A a las 8 horas de iniciado el tratamiento anterior.
Trasplante de células precursoras hematopoyéticas.
Estudio HLH-2004: 8
Variación principal: La ciclosporina A se administra desde el inicio del tratamiento.
Las guías de tratamiento están dirigidas a contener el fracaso medular al inhibir la hemofagocitosis; de esto resulta que el tratamiento inicial se haga sobre la base de corticoides e inmunoglobulinas, que pueden mejorar significativamente el pronóstico de esta enfermedad. Sin embargo, existe otro tipo de medida terapéutica, como por ejemplo, la administración de factores estimulantes de colonias de granulocitos, que se han utilizado en otros países en pacientes con síndrome hemofagocítico asociado a infección viral, que obtuvo muy buenos resultados. En términos generales esta paciente llevaba varios meses de episodios febriles aislados sin otra sintomatología, tratada por diferentes facultativos por anemia, constatada en una serie hemogramas realizados.
La paciente no era portadora de ninguna enfermedad, ni consumidora de medicamentos, salvo la ingestión de Herbalife© para bajar de peso. Es importante destacar que en este caso, el primer síntoma premonitorio de la enfermedad fue un síndrome febril prolongado, refractario al tratamiento antitérmico convencional. Otro hallazgo relevante desde el inicio de la enfermedad, fue la presencia de bicitopenia (anemia normocítica, leucopenia). Lo que hizo plantear inicialmente la posibilidad de un proceso autoinmune (LES), leishmaniosis visceral (LV), o una infección por HIV, por tratarse, de una mujer joven, sexualmente activa.
Estas consideraciones demuestran las dificultades iniciales que plantean estos enfermos, en donde la sintomatología puede ser explicada por estas patologías, que posteriormente fueron descartadas en nuestra paciente. Se pueden confundir fácilmente estas entidades y por lo tanto demorar el diagnóstico de la enfermedad.
En los pacientes con citopenia de 2 o más linajes celulares, la presencia de los parámetros como hiperferritinemia, coagulopatía, debe alertar ante la posibilidad de SAM, lo que debe considerarse tempranamente la realización de punción/aspiración de médula ósea y el hallazgo de hemofagocitosis. Esto acelera el diagnóstico y favorece el pronóstico. Se ha descrito que la biopsia de médula ósea es practicada para descartar infección o neoplasia, por lo que es considerada un método de estudio menos sensible que la aspiración de médula ósea.
Los hallazgos que el clínico debería tomar en cuenta para hacer diagnóstico de SAM son: fiebre persistente que no se explique por la condición de base (en el caso de tratarse de un síndrome hemofagocitico secundario), hepatoesplenomegalia y hemorragias. En el laboratorio se puede encontrar leucopenia, plaquetopenia o ambas, aumento de las enzimas hepáticas, hipertrigliceridemia, plaquetopenia, aumento de la ferritina sérica y de la deshidrogenada láctica. Sin embargo, lo más llamativo son las alteraciones de la coagulación, siendo la hipofibrinogenemia y el incremento de los niveles de ferritina sérica los hallazgos más importantes.
De esta manera, una de las características más relevantes del SAM lo constituye la presencia de una coagulación intravascular diseminada (CID), uno de los principales factores que condicionan la elevada mortalidad de este cuadro. Por esta razón es importante objetivar si existe prolongación del tiempo de protrombina, hipofibrinogenemia y presencia de productos de degradación de la fibrina.
La mayoría de las alteraciones descritas pueden explicarse por la infiltración histiocítica de los parénquimas. Las anomalías de la coagulación parecen ser causadas por la sobreposición de infiltración macrofágica a nivel hepático, lo que implica una disminución del fibrinógeno y de factores de coagulación dependientes de vitamina K. Además de una incapacidad de degradar factores procoagulantes y, por último la activación macrofágica que lleva a una liberación masiva de proteasas (que activan el plasminógeno) y liberación de TNF-a, que en grandes concentraciones, es capaz de inducir una coagulación intravascular diseminada.
Otro hallazgo interesante es el aumento de los niveles de triglicéridos, el que se relacionaría con la liberación de diversas citoquinas, especialmente TNF-a, que reduce la actividad de la lipoproteinlipasa. Cuando se logre documentar con mayor amplitud la utilización de marcadores tempranos de SAM, se podrá facilitar el trabajo del clínico en el tratamiento de este. Al respecto, existen estudios prometedores en el hallazgo de un marcador temprano: disminución marcada de ferritina glucosilada, pero esto se encuentra aún en estudio.
El elemento clave que orientó el diagnóstico de SAM fue el mielograma realizado en los últimos días de internación de la paciente en nuestro hospital, en el cual se constató la presencia de una intensa infiltración por elementos hemofagocíticos. Las causas desencadenantes de SAM en nuestra paciente fueron difíciles de establecer. Una inmunodeficiencia de base, no pudo confirmarse mediante los estudios pertinentes (Electroforesis de las proteínas, HIV negativo).
Los distintos estudios para pesquisar una enfermedad reumatológica fueron negativos y la presencia de una enfermedad linfoproliferativa fue descartada. La biopsia de médula ósea presentaba las tres series conservadas y diseritropoyesis marcada compatible con mielodisplasia secundaria con inmunomarcación negativa para linfoma.Sin embargo, hay que recalcar la falta de ciertos análisis como el estudio de anticuerpos específicos que no se practicaron por razones económicas. Por otra parte, la ausencia de antecedentes hereditarios hace poco probable que esta paciente fuera portadora de linfohistocitosis hemofagocítica familiar. No se pudo establecer una patología de base, pero sí la presencia de un proceso infeccioso bacteriano severo (neumonía), que pudo haber contribuido con la una falla orgánica múltiple.
En síntesis, la importancia de conocer este síndrome radica, en pensar esta patología en pacientes que comienzan con un proceso febril prolongado, cambios en la fórmula hematológica, que podría constituir los primeros signos de un SAM. Como se ha mencionado previamente, habitualmente el diagnóstico se establece de manera tardía, en varias ocasiones incluso post mortem, por la errónea presunción que se establece de que las fallas orgánicas son secundarias al proceso séptico.
Queda la duda razonable de si el SAM es el causante de la falla multiorgánica o si la superposición de infecciones en pacientes inmunodeprimidos propician que estos evolucionen hasta un desenlace fatal.
REFERENCIAS
1. Nuñez J, Montiel L, Nuñez del Prado JR. Síndrome hemofagocítico asociado infección viral por citomegalovirus. Medicina Intensiva. Barcelona. 2011;3(35):1-7. [ Links ]
2. Hadchouel M, Prieur A, Griscelli C. Acute hemorrhagic, hepatic and neurologic disease in juvenile rheumatoid arthritis. Possible relationship with drugs or infection. J Pediatr 1985;106:561-566. [ Links ]
3. Arceci R. When T cells and macrophages do not talk: The hemophagocytic syndromes. Curr Opin Hematol. 2008;15:359-367. [ Links ]
4. Kumakura S. Hemophagocytic syndrome. Intern Med. 2005;44:278-280. [ Links ]
5. Henter J, Horne A, Arico M, Egeler M, Goran E, Filipovich A, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. [ Links ]
6. Emmeneger U, Shaer D, Larroche C, Neftel K. Haemophagocytic syndrome in adults: Current concepts and challenges ahead. Swiss Med Wkly. 2005;135:299-314. [ Links ]
7. Fisman D. Hemophagocytic syndromes and infection. Emerg Infect Dis. 2000;6:601-608. [ Links ]
8. Filipovich L, Henter J, Janka G, Arico M, Ishii E. Histiocytosis Association of America. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. [ Links ]
9. Tuon F, Gomes V, Amato V, Graf M, Fonseca G, Lazari C, et al. Hemophagocytic syndrome associated with hepatitis A: Case report and literature review. Rev Inst Med Trop Sao Paulo. 2008;50:123-127. [ Links ]
10. Tsuda H, Shirono K. Successful treatement of virus-associated haemophagocytic syndrome in adult by cyclosporine A supported by granucyte colony-stimulating factor. Brit J Haemat. 1996;93:572-575. [ Links ]
11. Nogueira M, Vidal L, Terra B, Pagot T, Salluh J, Soares M. Hemophagocytic syndrome associated cytomegalovirus infection in a severely inmunocompromised AIDS patient: Case report. Braz J Inf Dis. 2009;13:72-73. [ Links ]
12. Kleinert M, Garate G, Osatnik J, Cicco J, Hunter B, Soria E. Síndrome hemofagocítico reactivo en pacientes graves, comunicación de 4 casos. Medicina (Buenos Aires). 2007;67:49-52. [ Links ]
13. Montiel L, Posadas C, Dominguez C. Fisiopatología del síndrome hemofagocítico (linfohistiocitosis hemofagocítica). Med Int Mex. 2005;21:75-81. [ Links ]
14. Arlet J, Le Thi Huong D, Marinho A, Amoura Z, Wechsler B, Papo T, et al. Ann Rheum Dis 2006;65:1596-1601. [ Links ]
15. Freeman H, Ramanan A. Review of haemophagocytic lymphohistiocytosis. Arch Dis Child 2011;96:688-693. [ Links ]
16. Fardet L, Coppo P, Kettaneh A, Dehoux M, Cabane J, Lambotte O. Low glycosylated ferritin, a good marker for the diagnosis of hemophagocytic syndrome. Arthritis Rheum. 2008;58:1521-1527. [ Links ]
17. Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn E. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients. A postmortem clinicopathologic analysis. Crit Care Med. 2004;32:1316-1321. [ Links ]
18. Gauvin F, Toledano B, Champagne J, Lacroix J. Reactive hemophagocytic syndrome presenting as a component of multiple organ dysfunction syndrome. Crit Care Med. 2000; 28:3341-3345. [ Links ]
*Correo electrónico: dradoramontiel@hotmail.com

