










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAnales de la Facultad de Ciencias Médicas (Asunción)
versão impressa ISSN 1816-8949
An. Fac. Cienc. Méd. (Asunción) vol.47 no.2 Asunción dez. 2014
ARTÍCULO ORIGINAL
Detección de la mutación JAK2V617F en pacientes con sospecha clínica de neoplasia mieloproliferativa sin expresión leucémica. Estudio preliminar.
JAK2V617F mutation detection in patients with clinical suspicion of myeloproliferative neoplasm without leukemic expression. Preliminary study.
Espínola Cano AF, Sánchez Mendoza S, Ayala Lugo A, Figueredo Thiel SJ
Hematopatología y Genética Molecular. Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción
RESUMEN
Se define como neoplasia mieloproliferativa a la alteración clonal de la célula madre hematopoyética caracterizada por la proliferación de uno o más linajes celulares mieloides en la médula ósea. La policitemia vera, la mielofibrosis primaria y la trombocitemia esencial son las entidades sin expresión leucémica más frecuentes en este grupo. La mutación puntual en el gen Januskinase 2 conocida como JAK2V617F, es una lesión génica encontrada en estas patologías. El objetivo de este estudio preliminar fue determinar la presencia de la mutación JAK2V617F en pacientes con sospecha clínico-patológica de neoplasia mieloproliferativa sin expresión leucémica utilizando la técnica de ARMS-PCR en los Laboratorios de Hematopatología y Genética Molecular del Instituto de Investigaciones en Ciencias de la Salud. En el 64% de pacientes (9 de 14 muestras de sangre periférica) se detectó la mutación. Su presencia constituye un valor diagnóstico sub-clasificatorio, pronóstico y terapéutico, siendo la técnica empleada de fácil ejecución e interpretación, obteniéndose resultados satisfactorios.
Palabras clave: neoplasia mieloproliferativa, JAK2V617F, ARMS-PCR.
ABSTRACT
Myeloproliferative neoplasms are clonal disorders of the haematopoietic stem cell characterized by the myeloid progenitors proliferation in bone marrow. Polycythemia vera, essential thorombocythemia and primary myelofibrosis are deseases without a leukemic condition. JAK2V617F is the denomination of an acquired point mutation found in the Januskinase 2 gene and associated to these pathologies. The aim of this preliminary study was to determinate the presence of JAK2V617F mutation in patient with suspected myeloproliferative disorders without leukemic condition using ARMS-PCR in the Hematopathology and Molecular Genetics Laboratory of the Instituto de Investigaciones en Ciencias de la Salud. In 64% of patients (9/14 blood samples) the mutation was detected. Detection of this mutation is useful in differential diagnosis, prognosis and therapeutic, furthermore, the assay is easy to perform and interpret.
Key words: myeloproliferative disorders, JAK2V617F, ARMS-PCR.
INTRODUCCIÓN
La policitemia vera (PV), la mielofibrosis primaria (MP) y la trombocitemia esencial (TE) se encuentran sub clasificadas dentro de un grupo de enfermedades, actualmente denominadas como neoplasia mieloproliferativa (NMP) por la Organización Mundial de la Salud (OMS) (1). Estas se definen como alteraciones clonales originadas a partir de la célula madre hematopoyética y caracterizadas por la proliferación de los linajes celulares mieloides entre los cuales se incluyen el linaje granulocítico, eritroide, megacariocítico y mastocítico. En el caso de la PV se observa un aumento de glóbulos rojos mediante mecanismos independientes a la regulación normal de la eritropoyesis, en tanto que en la MP se constata una proliferación de megacariocitos y granulocitos en la médula ósea. Por su parte, en la TE se verifica un aumento predominante del linaje celular megacariocítico. Otra patología importante, también incluida dentro de este grupo, es la Leucemia Mieloide Crónica (LMC) que constituye el prototipo del grupo de NMP (1).
Se ha demostrado que la alteración genética característica en la LMC es la aparición del gen de fusión BCR-ABL, producto de la translocación balanceada t(9;22) (q34;q11.2) a partir de la cual surge el cromosoma Philadelphia (Cr Ph) (2). Sin embargo, en las demás enfermedades incluidas como NMP, específicamente sin expresión leucémica, esta alteración se encuentra ausente, por lo cual se ha estimulado la búsqueda de otras posibles lesiones genéticas asociadas en éstas. Así se logra identificar una mutación puntual en el exón 14 del gen Januskinase 2 (JAK2) localizada en la región 9p24 del cromosoma 9, conocida como JAK2V617F. La misma se encuentra presente en el 95% de casos de PV y en 50% de casos de MP y TE (3).
Januskinase 2 es un miembro de la familia Janus de tirosina-quinasas citosólicas no receptoras, que incluye a JAK1, JAK3 y TYK2. La mutación JAK2V617F fue identificada en el año 2005 por varios grupos de investigación independientes, como una mutación recurrente en pacientes con NMP no leucémicas como PV, TE y MP (4-6).
La mutación descrita corresponde a una sustitución de una guanina por una timidina en el exón 14, posición 1849, que provoca el cambio del aminoácido valina por fenilalanina del codón 617 produciendo activación del dominio tirosin-quinasa. La misma fue detectada únicamente en células afectadas en 301 de 313 pacientes con NMP (96%), no así en las células de epitelio bucal. Esto sugiere que en la mayor parte de los casos se trata de una mutación somática adquirida por la célula madre hematopoyética, confiriéndole una ventaja proliferativa (7).
Para la detección de la mutación se puede recurrir al Sistema de Amplificación de Mutación Refractaria - Reacción en Cadena de la Polimerasa (ARMS-PCR por sus siglas en inglés). Este, permite la amplificación simultánea de las versiones normales y mutadas del gen, así como, la amplificación completa del Exón 14, cuyo producto se utiliza como control de calidad de la muestra y de la extracción del ADN, utilizando dos pares de cebadores en un único tubo de reacción de PCR. De esta manera se puede verificar si el paciente presenta o no la mutación y en caso de presentarla, si es heterocigoto u homocigoto y si la calidad del ADN extraído permite o no el estudio. Además, otra ventaja del método es que presenta una sensibilidad analítica del 0,1 al 0,05% (8), superior a otros métodos como la ASO-PCR de 1 al 3%, la PCR-RFLP de aproximadamente 4% (5,9) o la secuenciación de entre 20 y 30% (8,9).
El objetivo de este estudio preliminar fue determinar la presencia de la mutación JAK2V617F en pacientes adultos con sospecha clínica de neoplasia mieloproliferativa sin expresión leucémica mediante la técnica de ARMS-PCR, realizando además una descripción de las características demográficas y los hallazgos clínico-laboratoriales de los mismos y estimando la frecuencia de la mutación según su subclasificación clínico-morfológica en PV, MF o TE.
MATERIALES Y MÉTODOS
Población de estudio: Se realizó un estudio observacional descriptivo de corte transverso, preliminar (piloto), que incluyó a una población de 14 pacientes adultos, de ambos sexos, con sospecha clínica o diagnóstico anatomopatológico de neoplasia mieloproliferativa sin expresión leucémica. En estos se realizaron estudios citohistológicos y de patología molecular (detección de la mutación JAK2V617F) en los Laboratorios de Hematopatología y Genética Molecular del Instituto de Investigaciones en Ciencias de la Salud de la Universidad Nacional de Asunción (IICS-UNA). El estudio molecular fue realizado en muestras de sangre periférica extraídas por venopunción, empleándose la técnica denominada, Sistema de Amplificación de Mutación Refractaria (ARMS-PCR), para determinar la presencia o no de la mutación.
Extracción de ADN: Las muestras de sangre periférica fueron colectadas en tubos estériles con anticoagulante EDTA. La extracción de ADN se realizó utilizando el kit Wizard Genomic DNA Purification (Promega, USA) siguiendo las especificaciones del fabricante. La concentración de DNA necesaria para la PCR es de 0,1 µg/µL el cual es mantenido en heladera a 4°C hasta el momento de realizar la determinación y posteriormente almacenado en freezer a -65°C.
PCR: El DNA genómico fue utilizado para realizar el ARMS-PCR según el método descrito por Jones y col (8), modificado por Chen y col (10), para el cual se utilizaron cuatro cebadores (primers), un par directo/reverso (FO/RO) específicos para el exón 14 del gen JAK2, uno directo específico para la detección de la versión silvestre (no mutante) del gen (Fwt) y otro reverso para la detección de la versión mutante (Rmt). Las secuencias de dichos primers fueron: FO, 5' TCCTCAGAACGTTGATGGCAG 3'; RO, 5'ATTGCTTTCCTTT- TTCACAAGAT 3'; Fwt, 5'GCATTTGGTTTTAAATTATGGAGTATaTG 3' y Rmt, 5'GTTTTACTTACTCTCGTCTCCACAaAA 3'.
Los nucleótidos escritos en minúsculas representan desapareamientos intencionales que maximizan la discriminación entre alelos y los subrayados indican el nucleótido genotipo-específico.
Las PCR fueron realizadas en un volumen reacción de 50 µL empleando 100 ng de DNA genómico, 0,4; 0,3; 0,5 y 1,0 µmol/L de los primers FO, RO, Fwt y Rmt respectivamente, 120 µmol/L de dNTPs, 1,5 mmol/L de MgCl2 y 2 U de DNApolimeraza AmpliTaq Gold (Applied Biosystems, USA). Las amplificaciones se realizaron en un termociclador Esco Healthcare Swift MaxPro (China) por 40 ciclos. Cada ciclo consistió en 30s de desnaturalización a 94°C, 45s de templado a 58°C y 45s de extensión a 72°C.
El periodo de extensión final fue de 10 min a 72°C. Como paso previo se realizó un calentamiento a 95°C por 10 min para la activación de la DNA polimerasa.
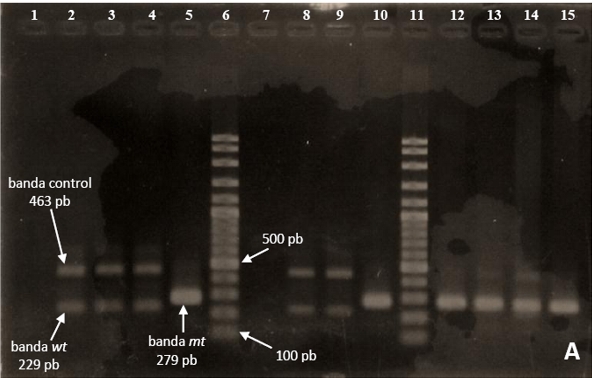
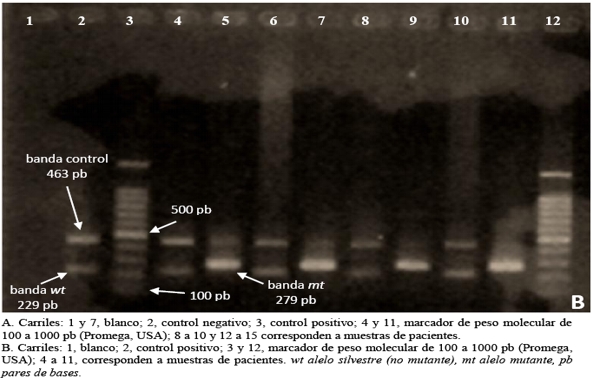
Visualización de los productos PCR: Los amplicones fueron resueltos con electroforesis en gel de agarosa al 2% revelado con bromuro de etidio o SYBR Safe (Invitrogen, USA). Los productos de PCR específicos muestran las siguientes bandas: 463 pb correspondiente al exón 14 del gen JAK2 que funge de control interno de amplificación, 279 pb que indica la presencia del alelo mutante y 229 pb corresponde al alelo silvestre (no mutante).
Control de calidad de la PCR: Los experimentos incluyeron además de las muestras de interés un blanco de reacción consistente en agua destilada, un control negativo obtenido de un donante sano y un control positivo proveniente de un paciente con policitemia vera positivo para la mutación JAK2V617F.
Para las características demográficas y los hallazgos clínico-laboratoriales se contó con un instrumento a modo de cuestionario en el que se incluyeron todos los datos relevantes de cada paciente.
Asuntos éticos: Se contó con la aprobación del comité de ética del Instituto de Investigaciones en Ciencias de la Salud para el protocolo del trabajo. Cada participante firmó una hoja de consentimiento informado. Además, se respetó la confidencialidad de los datos brindados y los resultados obtenidos de cada paciente.
RESULTADOS
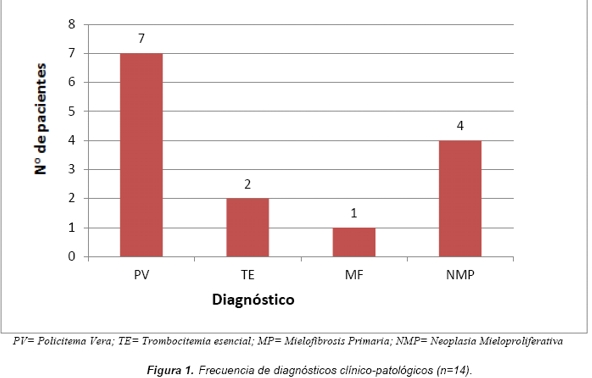
Se estudiaron 14 pacientes con sospecha clínica o diagnóstico anatomopatológico de neoplasia mieloproliferativa sin expresión leucémica, 10 de los cuales fueron del sexo femenino y 4 del sexo masculino con rango de edad comprendido entre 32 y 74 años. Siete pacientes presentaron diagnóstico de policitemia vera, dos de trombocitemia esencial y uno de mielofibrosis primaria. Los cuatro restantes concurrieron como neoplasia mieloproliferativa sin sub-clasificar Figura 1.
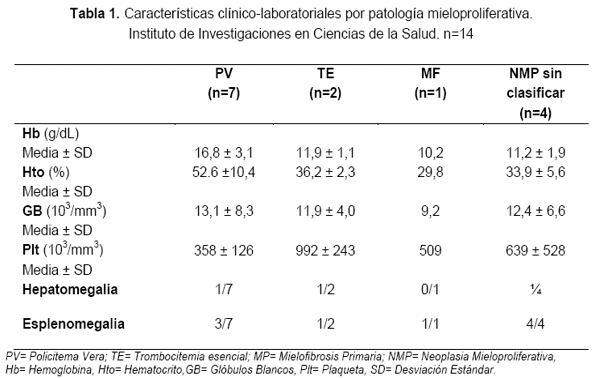
En cuanto a la características clínicas de la población estudiada, resumidas en la Tabla 1, se observó en los casos de policitemia vera un notable aumento en la hemoglobina (Hb) y el hematocrito (Hto), medias de 16,8 ± 3,1 g/dL y 52,6 ±10,4 % respectivamente.
Tres casos presentaron leucocitosis, presentando los demás, valores dentro de los parámetros normales. En todos los casos de PV el recuento plaquetario estuvo dentro de los valores normales. Se evidenció hepatoesplenomegalia en uno de los pacientes y esplenomegalia en otros dos.
En los dos pacientes con trombocitemia esencial, se observaron valores dentro de parámetros normales para hemoglobina y hematocrito, sin embargo uno de ellos presentó leucocitosis moderada, trombocitosis por encima de las 800.000 plaquetas/mm3 en ambos casos y además uno de ellos presentó hepatoesplenomegalia.
Un solo paciente presentó mielofibrosis primaria con ligera anemia, recuento leucocitario normal y trombocitosis, además de esplenomegalia.
Por último, en los casos sospechosos de neoplasia mieloproliferativa sin sub-clasificar, se observaron valores de hemoglobina y hematocrito ligeramente disminuidos con medias de 11,9 g/dL y 36% respectivamente, además de leucopenia, leucocitosis y trombocitosis.
Solo en siete pacientes se realizaron estudios de genética molecular para el transcripto BCR-ABL1 los cuales resultaron negativos, cuatro de ellos presentaron además estudios de cariotipo sin reportarse anomalías cromosómicas estructurales ni numéricas.
En cuanto a los resultados de ARMS-PCR para la mutación JAK2V617F, el 62% (9/14) de los pacientes presentó la mutación, la cual se ilustra en las figuras 2 A y B.
De los pacientes con diagnóstico anatomopatológico y/o sospecha clínica de policitemia vera, el 86% (6/7) resultaron positivos, para los casos de trombocitemia esencial el 100% (2/2) fue positivo y en el único caso de mielofibrosis primaria no se detectó la mutación. En los cuatro pacientes con diagnóstico de síndrome mieloproliferativo sin sub-clasificar el 25% (1/4) resultó positivo (Tabla 2).

DISCUSIÓN
Este proyecto fue un estudio preliminar sobre las enfermedades que se incluyen actualmente como neoplasia mieloproliferativa sin expresión leucémica por lo que la cantidad de pacientes reclutados no es suficiente para un análisis estadístico exhaustivo. Sin embargo resultó de suma utilidad para implementar y estandarizar la técnica de ARMS-PCR para el estudio genético molecular como ayuda al diagnóstico (3-6). Esta técnica presenta la ventaja de ser un método sencillo, fácil y reproducible para detectar la presencia de la mutación JAK2V617F en pacientes con neoplasia mieloproliferativa y poder así subclasificar junto con los hallazgos clínicos y morfológicos en las diferentes patologías que incluye este grupo (1).
Así, la mayor parte de los pacientes estudiados (7/14) correspondieron a casos de policitemia vera siendo detectada la mutación JAK2V617F en el 86% (6/7), una proporción similar a la encontrada por otros investigadores y citada comúnmente en la literatura donde se cita que en entre 90 y 95% de las policitemia vera presenta la mutación, al igual que aproximadamente el 50% de las trombocitemia esencia y la mielofibrosis primaria. En este trabajo si bien no se correlaciona completamente estas proporciones obtenidas, sería debido a la escasa cantidad casos de este estudio preliminar. No obstante se aprecia que para los casos más frecuentes como la PV se presenta una relación similar (1). En cuanto a las demás patologías de este grupo, como ser la TE y MP, el número estudiado fue aún muy pequeño como para contrastarlos (7). Cabe destacar también, que cuatro participantes acudieron a realizarse el estudio con neoplasia mieloproliferativa sin sub-clasificar, esto se debe probablemente a que los datos clínicos fueron insuficientes y no se realizaron todos los estudios laboratoriales complementarios antes del estudio genético molecular. De esto se deduce la importancia de realizar el diagnóstico multidisciplinario conjunto en este grupo de patologías.
AGRADECIMIENTOS
A la Dirección General de Investigación Científica y Tecnológica de la Universidad Nacional de Asunción por el financiamiento del proyecto.
REFERENCIAS BIBLIOGRÁFICAS
1. Swerdlow SH, Vardiman JW, Campo E, Harris NL, Jaffe ES, Pileri SA, et al., editores. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Cuarta Edición. Lyon: IARC; 2008. 439 p [ Links ]
2. Melo JV, Hughes TP, Apperley JF. Chronic Myeloid Leukemia.American Society of Hematology Education Program Book. 2003 Ene;(1):132–52. [ Links ]
3. Tefferi A, Skoda R, Vardiman JW. Myoloproliferative neoplasms: contemporary diagnosis using histology and genetics. Clinical Oncology. 2009 Nov;6:627–37. [ Links ]
4. James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythemia vera. Nature. 2005 Abr;434:1144–8. [ Links ]
5. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005 Abr;7:388–97. [ Links ]
6. Kralovics R, Passamonti F, Buser AS, Soon-Siong T, Tiedt R, Passweg JR, et al. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. New England Journal Of Medicine. 2005 Abr;352(17):1779–90. [ Links ]
7. Levine RL, Pardanani A, Tefferi A, Gilliand DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nature. 2007 Sep;7:673–83. [ Links ]
8. Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of tha JAK2 V617F mutation in chronic myeloproliferative disorders. BLOOD. 2005 Sep;106(6):1162–8. [ Links ]
9. Campbell PJ, Green AR. The Myeloproliferative Disorders. New England Journal Of Medicine. 2006 Dic;355(23):2452–66. [ Links ]
10. Chen Q, Lu P, Jones AV, Cross NCP, Silver RT, Wang YL. Consultation in Molecular Diagnostics: Amplification Refractory Mutation System, a Highly Sensitive and Simple Polymerase Chain Reaction Assay, for the Detection of JAK2 V617F Mutation in Chronic Myeloproliferative Disorders. JMD. 20079(2):272-276. [ Links ]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}