










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAnales de la Facultad de Ciencias Médicas (Asunción)
versão impressa ISSN 1816-8949
An. Fac. Cienc. Méd. (Asunción) v.46 n.2 Asunción dez. 2013
REPORTE DE CASO
Síndrome de Morris. Reporte de un caso
Morris Syndrom. Case report
Acuña Apleyard V, Wildberger Ramirez C, Espínola Castiglioni R.
Cátedra de Ginecología y Obstetricia. Facultad de Ciencias Médicas.
Universidad Nacional de Asunción
RESUMEN
El Síndrome de Insensibilidad Androgénica es una forma de seudohermafroditismo masculino en la que los órganos diana son incapaces de reaccionar a la testosterona o dihidrotestosterona. Como consecuencia, aunque las gónadas son testículos, los genitales no se virilizan adecuadamente durante el desarrollo fetal. El cuadro clínico varía desde pacientes con genitales femeninos en la resistencia androgénica completa o síndrome de Morris; genitales ambiguos en la resistencia parcial o síndrome de Reifenstein; hasta genitales masculinos normales en los casos de formas más ligeras de la resistencia androgénica.
Se presenta el caso de una mujer adulta que consulta a la Cátedra y Servicio de Ginecología y Obstetricia de la Facultad de Ciencias Médicas de la Universidad Nacional de Asunción en agosto de 2012. La misma acude por deseo de mejor calidad de vida sexual. La paciente se conoce portadora de Síndrome de Morris desde el año 2006. Se realiza tratamiento quirúrgico para ampliación vaginal obteniéndose resultados favorables constatados durante el seguimiento de la paciente.
Palabras clave: Síndrome de Morris, amenorrea, insensibilidad androgénica.
ABSTRACT
Androgen Insensitivity Syndrome is a form of male pseudohermaphroditism in which the target organs are unable to respond to testosterone or dihydrotestosterone. As a result, although the gonads are testes, genitalia virilization do not properly occurs during fetal development. The clinical manifestations varies from female genitalia in patients with complete androgen resistance or Morris syndrome; ambiguous genitalia in partial resistance or Reifenstein syndrome; to normal male genitalia in cases of milder forms of androgen resistance.
This is a case report of an adult woman who is attended at the Service of Obstetrics and Gynecology of the School of Medicine of the Universidad Nacional de Asunción. She wishes for a better sexual life. The patient is known to have Morris syndrome since 2006. Surgery is performed to expand the vagina, obtaining favorable results as it was confirmed in the follow up.
Key words: Morris Syndrom, amenorrhea, androgenic insensitivity.
INTRODUCCION
La amenorrea es la ausencia de la menstruación. Se considera primaria si no se ha producido la menarquia a los 16 años de edad y secundaria en aquellos casos con ausencia de menstruación por un periodo igual o mayor a tres meses, en mujeres que han menstruado con anterioridad. La ausencia de la menstruación refleja una alteración en el eje gonadal o en las vías genitales y puede ocurrir en mujeres con fenotipo normal, asociarse a hipogonadismo, disgenesia gonadal o a síntomas de hiperandrogenismo (1).
El Síndrome de Insensibilidad Androgénica (SIA) es una forma de seudohermafroditismo masculino en la que los órganos diana son incapaces de reaccionar a la testosterona (T) o dihidrotestosterona (DHT). Como consecuencia, aunque las gónadas son testículos, los genitales no se virilizan adecuadamente durante el desarrollo fetal (2,3). El cuadro clínico varía desde pacientes con genitales femeninos en la resistencia androgénica completa o síndrome de Morris; genitales ambiguos en la resistencia parcial o síndrome de Reifenstein; hasta genitales masculinos normales en los casos de formas más ligeras de la resistencia androgénica (4).
El síndrome de resistencia completa a los andrógenos, feminización testicular o síndrome de Morris, puede presentarse en uno de cada 20.000 a 64.000 recién nacidos varones. Se lo considera la tercera causa de amenorrea primaria, después de la disgenesia gonadal y la ausencia congénita de vagina. El síndrome tiene gran heterogeneidad genética, puede producirse por alteraciones cuantitativas o cualitativas del receptor androgénico y se caracteriza por el fenotipo femenino en individuos 46 XY (5).
El diagnóstico se establece generalmente cuando el individuo acude por una hernia inguinal antes de la pubertad o por una amenorrea primaria después de la pubertad. Los genitales externos son femeninos pero con hipoplasia de los labios mayores y menores. La vagina es poco profunda y termina en un fondo de saco ciego. Las mamas tienen buen desarrollo.
Los testículos se localizan en el abdomen, canal inguinal o labios mayores y son normales antes de la pubertad. Es frecuente la presencia de carcinoma in situ y de seminoma testicular después de la pubertad, por lo que es recomendable la extirpación profiláctica de los mismos (6).
CASO CLINICO
Paciente de 32 años que consulta a la Cátedra de Ginecología y Obstetricia de la Facultad de Medicina de la Universidad Nacional de Asunción. La misma refiere deseo de mejorar su vida sexual. Antecedentes patológicos personales: conocida portadora de agenesia uterina desde el año 2006. Refiere hernia inguinal izquierda a los nueve meses de vida, con extirpación de ovario izquierdo. Presenta cariotipo 46XY. Antecedentes gineco-obstétricos: niega menarca. Niega gestas.
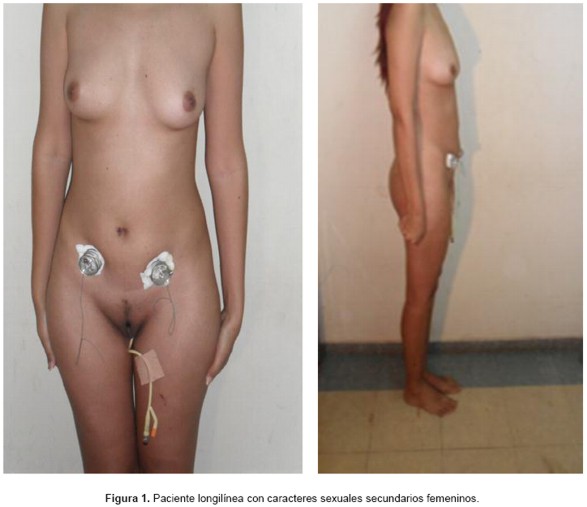
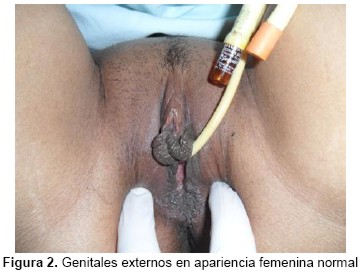
Examen Físico: apariencia longilínea. Mamas: simétricas, no se palpan nódulos. Abdomen: plano, simétrico, blando, depresible, no doloroso. Distribución pilosa femenina. Genitales externos de características femeninas normales (Figuras 1 y 2). Especuloscopía: cúpula vaginal normal, vagina corta (4-5 cm de longitud). Tacto vaginal: vagina corta.
Métodos auxiliares de diagnóstico: Ecografía de abdomen superior normal. Se observan ambos riñones normales. Laboratorio: FSH: 2.5 UI/ml (N: 3-20 UI/ml) Estradiol: 55ng/l (N: 30–300ng/l) Prolactina: 7.4µg/l (N: 5-25µg/l) TSH: 1.9mUI/l (N: 0,4-4mU/l) T4: 1.1ng/dL (N:08–4ng/dl). Ecografía transvaginal: Útero de 26x10x19mm difícil de visualizar. Interfase endometrial de 1mm. Ovarios derecho e izquierdo no caracterizados. Ecografía mamaria normal.
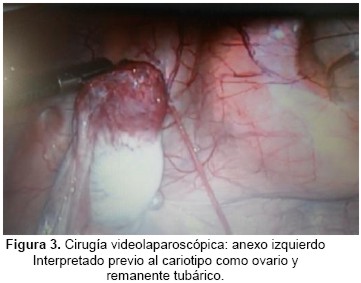
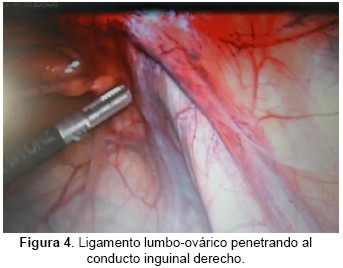
Se realiza cirugía vídeo laparoscópica diagnóstica en la que se constata: Ovario derecho ausente. Ligamento lumbo-ovárico que penetra en orificio inguinal profundo. Ovario Izquierdo presente. (Figura 3 y 4).
Se realiza biopsia del mismo y una punción aspirativa con aguja gruesa de un nódulo solido elástico de 4x3cm de diámetro ubicado en la región inguinal derecha. El informe de anatomía patológica revela en el material de punción fondo proteináceo sin atipias celulares. Biopsia de ovario Izquierdo: Estructuras microglandulares de escaso calibre separados por estroma fibroso, sin atipias, cuyo aspecto evoca características observadas en el tumor de tipo Sertoli. La paciente se niega a extirpación de los tumores a pesar de la explicación de la posibilidad de malignización de los mismos. Con tales resultados se procede a la ampliación vaginal quirúrgica mediante la fijación de la cúpula vaginal con hilos, a los accesos guía de video-laparoscopia y tracción diaria de los mismos por 12 días. Al cabo de este tiempo se retiran hilos por vía vaginal. Se realiza nuevo examen genital y se constata introito que deja penetrar dos dedos ampliamente y fondo más de un dedo que antes. Se coloca un espéculo mediano sin dificultad.
Se indica estrógenos locales por 15 días.El nuevo examen a los 15 días revela pasaje de dos dedos sin dificultad al tacto.
Forzando la vagina se amplía el fondo (Figura 5). Actualmente la paciente se encuentra con relaciones sexuales satisfactorias.
DISCUSION
El síndrome de insensibilidad andrógena o síndrome de Morris (antes llamado «feminización testicular») es un trastorno genético caracterizado por la escasa respuesta a los andrógenos (testosterona). Éste es un síndrome que se produce por una mutación en el gen receptor de andrógeno en el cromosoma X, por lo cual su herencia se describe como «recesiva ligada al X». Las madres portadoras del gen tienen un 50% de posibilidades de tener hijos con síndrome de insensibilidad androgénica o hijas con un 50% de posibilidades de ser portadoras de la mutación (7). Las personas afectadas son cromosómicamente masculinas (46XY) con genitales externos aparentemente femeninos, con vagina ciega, sin útero ni ovarios. Los testículos están presentes en el abdomen o en el canal inguinal y muchas veces son confundidos con hernias en los niños con apariencia fenotípica femenina normal (8).
El tratamiento para los trastornos de la diferenciación sexual dependerá de su tipo específico, pero se suele realizar una cirugía correctiva para resecar o crear los órganos reproductores apropiados para el sexo del paciente. El tratamiento también puede incluir la terapia de reemplazo hormonal (9). En el caso reportado la paciente conociendo la patología solicitó ser sometida a cirugía a fin de mejorar su calidad de vida sexual. Sin embargo y a pesar de las informaciones brindadas acerca de la posibilidad de malignización testicular, esta se negó a la extirpación del mismo, comprometiéndose a controles periódicos.
La correcta determinación del sexo es importante no sólo en lo que respecta al tratamiento sino también en lo relacionado con el bienestar emocional del niño. Estos pacientes deben ser criados en un ambiente donde se adapte con toda convicción al sexo «asignado» (9). La paciente en cuestión fue criada desde un principio como niña por lo cual la misma a pesar de conocer y entender su condición genética se sintió identificada con el sexo femenino desde un principio. Se realizó igualmente acompañamiento psicológico en el servicio, tanto para la paciente como para los familiares, a fin de entender las implicancias de la cirugía, así como sus limitaciones para su vida reproductiva.
El tratamiento y la asignación de un género son situaciones bastante complicadas por lo tanto deben ser abordadas en forma muy individualizada y con gran cuidado en cada caso. Se requiere de un conocimiento pleno de la patología como tal y en casos como este en el que la paciente ya tiene una identidad de género respetar independientemente del género cromosómico.
En pacientes con insensibilidad parcial a los andrógenos y genitales externos predominantemente femeninos, la literatura recomienda realizar gonadectomía antes de la pubertad, a fin de evitar disconfort y cliteromegalia cuando se alcance esta etapa. En aquellos pacientes con genitales ambiguos o predominantemente masculinos el proceso de asignación de sexo es más complejo. Además de considerar los aspectos puramente anatómicos y quirúrgicos es necesario realizar pruebas terapéuticas con testosterona a fin de predecir la potencial respuesta androgénica en la pubertad (10).
REFERENCIAS BIBLIOGRÁFICAS
1. Borrego J, Varona J, Areces G, Formoso L. Sindrome de Morris. Revista Cubana de Obstetricia y Ginecología. 2012; 38(3)415-423. [ Links ]
2. Akin JW, Behzadian A, Tho SPT. Evidence for a partial deletion in the androgen receptor gene in a phenotypic male with azoospermia. Am J Obstet Gynecol. 1991;165:1891. [ Links ]
3. Balducci R, Ghirri P, Brown TR, Bradford S. Clinician looks at androgen resistance. Steroids. 1996;61:205. [ Links ]
4. Marcelli M, Tilley WD, Wilson CM. A single nucleotide substitution introduces a premature termination codon into the androgen receptor gene of a patient with receptor-negative androgen resistance. J Clin Invest. 1990;85:1522. [ Links ]
5. Grumbach MM, Conte FA. Disorders of sex differentiation. In: Willsom JD, Foster DW, Kronrnberg HM, Larsen PR, editors. Williams Textbook of endocrinology. 9th Edition. W. B. Philadelphia: Saunders Co; 1998. p. 1303. [ Links ]
6. Mejias Y, Duani O, Taboada N. Trastornos de la diferenciación sexual: presentación de un caso de genitales ambiguos y revisión del tema. Rev Cubana Pediatr. 2007;79(3). [ Links ]
7. Osorio V, Alonso F.Sindrome de Feminizacion testicular incompleta. Arch. Esp. Urol., 2006; 59, 2 (179-182). [ Links ]
8. Sepulveda Agudelo J, Alarcon M, Jaimes H. Amenorrea primaria. Rev col. Obstet Ginecol, 2009: 60(1): 57-67. [ Links ]
9. Nowier A, Esmat M, Hamza RT. Surgical and Functional Outcomes of Sigmoid Vaginoplasty Among Patients With Variants Of Disorders of Sex Development. 2012 Vol. 38 (3): 380-388. [ Links ]
10. Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A. Androgen insensitivity syndrome: clinical features and molecular defects HORMONES 2008, 7(3):217-229. [ Links ]


{kind=link}
{kind=link}
{kind=link}
{kind=link}