










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Facultad de Ciencias Médicas (Asunción)
Print version ISSN 1816-8949
An. Fac. Cienc. Méd. (Asunción) vol.38 no.4 Asunción Dec. 2005
TRABAJO ORIGINAL
Síndrome uveo meníngeo de Vogt-Koyanagi-Harada. A propósito de un caso
Vogt-Koyanagi-Harada. Syndrome. A case report
Dr. Agustín Carron Alvarado1, Dra. Ingrid Castro2, Dr. Pablo Cibils3, Prof. Dr. Luis Ayala Haedo4.
1) Residente. Cátedra de Oftalmología. Hospital de Clínicas. FCM, UNA.
2) Fellow del Departamento de Retina. Cátedra de Oftalmología. Hospital de Clínicas. FCM, UNA.
3) Médico agregado. Cátedra de Oftalmología. Hospital de Clínicas. FCM, UNA.
4) Jefe de Cátedra y Servicio. Jefe del Departamento de Retina. Cátedra de Oftalmología. Hospital de Clínicas. FCM, UNA.
RESUMEN
Objetivo: Reportar un caso de Síndrome de Vogt-Koyanagi-Harada y describir las características clínicas del cuadro.
Diseño: Reporte de caso único, intervencionista
Métodos: Presentación de las características clínicas del caso.
Resultados: Se presenta un caso de Sx. de Vogt-Koyanagi-Harada en una paciente de 25 años de edad, tratada con corticoides a altas dosis, sin mejoría de la AV a pesar del tratamiento.
Conclusión: El Sx. de Vogt-Koyanagi-Harada es una panuveítis granulomatosa bilateral con comprometimiento auditivo, de las meninges y de la piel, la pérdida de la agudeza visual asociada es moderada a severa.
SUMMARY
Purpose: To report a case of Vogt-Koyanagi-Harada syndrome and describe the main clinical characteristics of the disease.
Design: Single interventional case report.
Methods: Case report and description of the clinical findings of the case.
Results: We present a 25 year-woman with Vogt-Koyanagi-Harada syndrome, treated with high doses of corticosteroids, without improvement of the visual acuity instead the treatment.
Conclusion: Vogt-Koyanagi-Harada disease is a granulomatous panuveitis that involves the eyes, auditory system, meninges, and skin. The loss of visual acuity is moderate to severe.
INTRODUCCIÓN
Los síndromes uveo-meníngeos son un grupo heterogéneo de enfermedades de diversa etiología que tienen en común el comprometimiento de la uvea, la retina y las meninges (1). Dentro de este variado grupo de patologías se encuentra en Sx. de Vogt-Koyanagi-Harada. (Tabla nº 1)
Se define entonces al Sx. de Vogt-Koyanagi-Harada como una panuveítis granulomatosa bilateral asociada a desprendimiento exudativo de la retina que se acompaña de manifestaciones cutáneas y neurológicas características (1,2,6).

REPORTE DEL CASO
Paciente de 25 años, de sexo femenino consulta a la Cátedra de Oftalmología del Hospital de Clínicas con historia de disminución progresiva de la agudeza visual (AV) en ambos ojos (AO) de aproximadamente un mes de evolución; acompaña al cuadro hipoacusia de instalación gradual; cefalea frontal de moderada intensidad y dolor retroocular. Nauseas y vómitos en 2 oportunidades en las últimas 24 horas. Hallazgos del examen ofltalmológico AV: Percepción luminosa con proyección en todos los cuadrantes en AO Datos laboratoriales Hemograma: dentro de límites normales Estudios de imágenes TAC: Sin alteraciones a nivel craneoencefálico, engrosamiento retiniano bilateral Evolución Se llega al diagnóstico de Síndrome de Vogt-Koyanagi-Harada, luego de haber descartado además posibles causas de hipertensión endocraneana. Se inicia tratamiento tópico con fosfato de dexametasona al 1% (1 gota cada 4 horas) + atropina (1 gota cada 8 horas). Se solicita internación en la Primera Cátedra de Clínica Médica para iniciar corticoterapia parenteral a altas dosis (bolos de Metil-prednisolona) 1g/día x 3 días; luego prednisona vía oral 75 mg/día. DISCUSIÓN Este caso corresponde a la denominada forma incompleta del Sx. de Vogt-Koyanagi-Harada, según los criterios revisados por el primer grupo de trabajo internacional sobre Sx de Vogt-Koyanagi-Harada; debido a que no presenta los cambios cutáneos característicos (Tabla nº 2) (7). CONCLUSIÓN El Sx de Vogt-Koyanagi-Harada es una panuveítis bilateral que además compromete el sistema auditivo, las meninges y la piel; por esta razón debe ser conocido no sólo por el oftalmólogo sino por los demás profesionales a quienes puede presentarse un paciente con estos síntomas. Los corticoides constituyen la base del tratamiento. El pronóstico visual depende de la edad de inicio, del tratamiento instaurado y de la aparición de complicaciones como fibrosis subretiniana o neovascularización coroidea que causan disminución irreversible de la AV. BIBLIOGRAFÍA 1. Brazis P, Stewart M, Lee A. The Uveo-Meningeal Syndromes. The Neurologist 2004; 10: 171–184. [ Links ]
Presión intraocular: 14 mmHg AO
Biomicroscopia: AO:
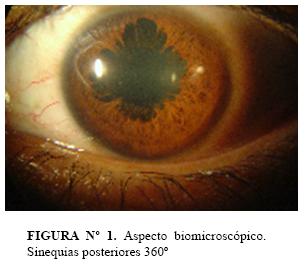
Congestión conjuntival y periquerática marcada, cornea transparente con precipitados queráticos finos, cámara anterior de profundidad normal, reacción de cámara anterior 4+/4+; sinequias posteriores que interesan los 360º del margen pupilar, cristalino transparente, vitreo muy turbio que solo deja ver papila congestiva AO; e impresiona desprendimiento exudativo de la retina (figura nº1).
Química sanguínea: dentro de límites normales
VDRL: reactiva 1:8
Análisis del LCR: límpido, incoloro
Glucosa: 0,45g/l; Proteínas: 0,30 g/l
Hematíes: 20/mm3
Leucocitos: 4/mm3
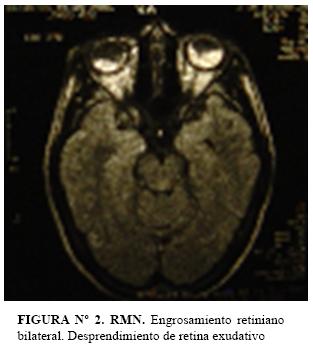
RMN: Dentro de límites normales; a excepción de marcado engrosamiento retiniano bilateral. (Figura nº 2)
Con el tratamiento tópico y sistémico la inflamación del segmento anterior y posterior del ojo mejoró notablemente, no así la AV debido a que no hubo mejoría alguna en el estado del desprendimiento de retina; por lo que se maneja la posibilidad de hacer inyección intravítrea de Triamcinolona.
El cuadro clínico de esta entidad simula en cierta forma un Síndrome de hipertensión endocraneana; de ahí la importancia que tanto el oftalmólogo como el clínico general tenga presente este cuadro relativamente raro; pero bastante característico a la hora de generar diagnósticos diferenciales.
El cuadro clínico se divide en cuatro fases (1,2,3,4,5).
Prodrómica: con manifestaciones de un Sx gripal, a las que se agregan meningismo, disacusia, alteración del estado de conciencia.
Uveítica: Iridociclitis granulomatosa, vitritis, congestión del nervio óptico y desprendimiento exudativo de retina.
Convalescencia: Remisión del cuadro inflamatorio ocular y aparición de las manifestaciones cutáneas.
Crónica-recurrente: Con ataques recurrentes de uveítis anterior severa, en esta fase aparecen la mayoría de las complicaciones oculares.
La etiología del cuadro es desconocida sin embargo varios investigadores sugieren su asociación con un proceso autoinmune o infeccioso; la presencia del HLA-DR1 y DR4 se encuentra en 85% de los casos (1,2,4,5,8).
Se considera que existe una reacción autoinmune mediada por linfocitos T contra un antígeno asociado a los melanocitos, el mecanismo desencadenante de la reacción también es desconocido pero podría ser una lesión cutánea o una infección viral.
El tratamiento se basa en la corticoterapia a altas dosis, Metil prednisolona 1g/día por tres días y luego prednisona 1 a 1,5 mg/kg/día con el objetivo de frenar el proceso inflamatorio y evitar las complicaciones asociadas a éste; el tratamiento es prolongado y dura entre 6 a 9 meses, en algunos casos con dosis de mantenimiento crónicas para evitar la aparición de recurrencias (6,9,10).
Las complicaciones oculares se presentan en 50 % de los casos aproximadamente y la más frecuente es la aparición de Cataratas seguida por glaucoma, neovascularización coroidea y fibrosis subretiniana (6,9).
La agudeza visual en estos pacientes está entre 20/60 y 20/200 en el 90% de los casos, existiendo un 11% de pacientes que no presenta mejoría de la AV a pesar del tratamiento, son factores de mal pronóstico la edad avanzada y la mala AV al inicio del tratamiento (6,9).
2. Intraocular inflammation and uveitis. Basic and Clinical Science Course. Volumen 9. American Academy of Ophthalmology. 2003-2004. pg 200-203. [ Links ]
3. Chan C, Whitcupp S, Nussenblat R. Simpathetic Ophthalmia and Vogt-Koyanagi-Harada Syndrome. Volumen 4. Capítulo 51. Duanes Ophthalmology en CD-ROM. [ Links ]
4. Read R, Rao N, Cunningham Jr E. Vogt-Koyanagi-Harada disease. Current Opinion in Ophthalmology 2000, 11:437–442. [ Links ]
5. Rajendram R, Evans M, Rao N.Vogt-Koyanagi-Harada Disease. International Ophthalmology Clinics. 1995;35:69–86. [ Links ]
6. Vasudeo M, Biswas J, Ganesh S. Analysis of 87 Cases with Vogt-Koyanagi-Harada Disease. Japanese Journal of Ophthalmology 2000;44:296–301. [ Links ]
7. First International Workshop on Vogt-Koyanagi-Harada Disease. Revised Diagnostic Criteria for Vogt-Koyanagi-Harada Disease. Am J Ophthalmol 2001; 131:647–652. [ Links ]
8. Romayne Boyd S, Young S, Lightman S. Immunopathology of the Noninfectious Posterior and Intermediate Uveitides. Survey Of Ophthalmology 2001; 46:209–233. [ Links ]
9. Read R, Rechodouni A, Butani N, Johnston R, Labree L, Rao N. Complications and Prognostic Factors in Vogt-Koyanagi-Harada Disease American Journal of Ophthalmology 2001;131:599–606. [ Links ]
10. Andrade R, Muccioli C, Farah M, Nussenblatt R, Belfort Jr. R. Intravitreal Triamcinolone in the Treatment of Serous Retinal Detachment in Vogt-Koyanagi-Harada Syndrome. American Journal of Ophthalmology 2004;137:572–574. [ Links ]



{kind=link}
{kind=link}
{kind=link}
{kind=link}