










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAnales de la Facultad de Ciencias Médicas (Asunción)
versão impressa ISSN 1816-8949
An. Fac. Cienc. Méd. (Asunción) v.38 n.4 Asunción dez. 2005
MONOGRAFIA
Trombofilia hereditaria. Prevalencia de mutaciones en el factor II, V y en la Metiltetrahidrofolato Reductasa, en una población de donantes de sangre en Paraguay (1)
Hereditary trombofilia. Prevalence of mutations in factor II, V and in the Metiltetrahidrofolato Reductasa, in a population of donors of Blood of Paraguay
Imelda Martínez (2), Rosa Valdez (3) Carlos Azambuja (4) Nicolás Estrada (5)
1) Monografía presentada en la FCM - UNA
2) Profesora de Patología y Clínica Médica. FCM – UNA. Jefe de Departamento de Hepatología. II Cátedra de Clínica Médica.
3) Jefe del Banco de Sangre. Hospital de Clínicas.
4) Jefe de Laboratorio Genia, Genética Molecular. Montevideo, Uruguay.
5) Estudiante de la Facultad de Medicina. Montevideo, Uruguay. Laboratorio de Genética Molecular.
RESUMEN
La trombofilia hereditaria es una tendencia genéticamente determinada a desarrollar trombosis. Entre las causas más comunes pueden citarse deficiencias de ciertas proteínas de la coagulación como la Antitrombina, la proteína C, la proteína S. Y también ciertas mutaciones en los factores de la coagulación: Factor V Leiden, Protrombina G20210A, y mutaciones en la enzima Metiltetrahidrofolatoreductasa (MTHFR), entre otras. SUMMARY Trombofilia hereditary is a tendency genetically determined to develop trombosis. Between the most common causes can be mentioned, protein deficiencies certain of the coagulation, like Antitrombina, protein C, protein S, and also, certain mutations in the factors of the coagulation: Factor V Leiden, Protrombina G20210A, and mutations in the Metiltetrahidrofolatoreductasa enzyme (MTHFR), among others. INTRODUCCIÓN Este trabajo de investigación sobre algunos factores de trombofilia hereditaria, ha surgido con el propósito de averiguar la prevalencia de algunas mutaciones genéticas más frecuentes, en el factor II, en el factor V y en la enzima Metiltetrahidrofolatoreductasa (MTHFR). El estudio se ha llevado a cabo en una población sana, de doscientos donantes de sangre del Banco de sangre del Hospital de Clínicas. No hay antecedentes de investigación sobre el tema en nuestro hospital, sí en otro 1, en una muestra más pequeña. En otros países latinoamericanos, sí, se han hecho estudios de factores genéticos de trombofilia 2,3. Generalidades El desarrollo de los desórdenes trombóticos en los seres humanos es una de las causas más comunes de morbilidad y mortalidad en el mundo occidental 4. Recordemos que la enfermedad trombótica puede ser arterial y venosa. En la primera el flujo sanguíneo y la presión son más elevadas que en el sistema venoso, elementos básicos a tener en cuenta al hacer la distinción entre ambos tipos de trombosis. Además, son importantes diferencias a hacer notar, la composición del trombo (rico en plaquetas en el arterial, y en fibrina en el trombo venoso) y la presencia de daño en la pared vascular (ateromas). Sin embargo, la distinción no es absoluta, y hay, por supuesto, mecanismos comunes. La perturbación de la hemostasia es capital en toda trombosis, aún cuando ella difiera en su naturaleza por su localización. Las influencias del medio ambiente, transitorias o no, pueden jugar un rol importante en la perturbación de la hemostasia e incidir como factores de riesgo para el desarrollo de trombosis, ya sean arteriales o venosas. Este término, medio ambiente, es usado en su sentido amplio y comprende situaciones como, embarazo, parto, ingestión de hormonas, cirugía, dieta y uso de tabaco. También comprende desórdenes intercurrentes tales como diabetes melitus, hipertensión arterial, dislipidemia, hiperhomocisteinemia (adquirida), y cambios locales en la pared vascular. Un poco de historia: Desde hace algunos siglos era conocido el hecho de que alteraciones hereditarias de la coagulación, causaban tendencia a padecer hemorragias, por ejemplo, la Hemofilia A o B. Por el contrario, el conocimiento de las alteraciones hereditarias de la coagulación que producen trastornos trombóticos, es de apenas algunas décadas. OBJETIVOS Nos hemos propuesto los siguientes objetivos: MATERIAL Y MÉTODOS Este es un estudio de prevalencias, es decir, estudio de corte transversal, con algún componente analítico, y de selección secuencial. Se llevó a cabo en una población de donantes de sangre del Hospital de Clínicas en los meses de noviembre y diciembre del año 2001. Muestras de sangre Se trabajó con 200 muestras de sangre de los donantes que acudieron secuencialmente al Banco de Sangre del Hospital de Clínicas, entre los meses de noviembre y diciembre del año 2001. Estos donantes, como ya se dijo, reunieron los criterios de selección para el estudio. Y se siguieron estrictamente las normativas de procedimiento general de selección del donante de sangre, del Servicio de Medicina Transfusional del Hospital de Clínicas. Ver Anexo N° 1. Extracción de ADN Se utilizó el sistema Wizard (Genomic DNA Purification System) de Promega. Amplificación por PCR Se describe a continuación el método utilizado. Digestión enzimática A los efectos de distinguir los alelos mutados de los nativos, se utilizaron las enzimas de restricción Hind III para Factor II y Factor V y Hinf I para MTHFR. Visualización de los productos amplificados Los geles de poliacrilamida se forman por la polimerización de la acrilamida por acción de un agente entrecruzador ('cross-linking'), la bis-acrilamida en presencia de un iniciador y un catalizador. Como iniciador se utiliza TEMED (N,N,N,N'-tetrametilnediamina) y como iniciador el ión persulfato (S2O8-) que se añade en forma de persulfato amónico. Las soluciones de acrilamida se desgasifican pues el oxígeno es un inhibidor de la polimerización. Además, durante la polimerización se libera calor que podría provocar la formación de burbujas en el seno del gel. La velocidad de polimerización viene determinada por la concentración de persulfato (catalizador) y TEMED (iniciador). RESULTADOS Se analizaron 200 muestras de sangre. DISCUSIÓN Y COMENTARIOS En los factores estudiados en este trabajo, II, V y en la enzima MTHFR, se han encontrado mutaciones. Estas son las responsables del estado de hipercoagulabilidad en los individuos portadores. El descubrimiento y el estudio de las mutaciones genéticas que aumentan la predisposición del hombre a la trombosis, ha revolucionado el diagnóstico y el tratamiento de los individuos en riesgo. La prevalencia de estas mutaciones varía en distintas poblaciones según su caracterización étnica. CONCLUSIONES BIBLIOGRAFIA 1. Guggiari P de, Bizzachi J, Kordich L, Genoud V, Cohenca B. Prevalencia del Factor V Leiden, Factor II G20210A. Trabajo libre presentado en el VIII Congreso Paraguayo de Cardiología, octubre 1999, Asunción, Paraguay. [ Links ]
Los objetivos han sido: Identificar una de las mutaciones más frecuentes y su prevalencia, en el gen del Factor V, de la Protrombina, de la MTHFR, en una población de donantes de sangre del Paraguay.
Para este trabajo de investigación prospectivo y de corte transverso, se han tomado muestras de sangre de 200 donantes del Banco de Sangre del Hospital de Clínicas, que reunieron los criterios de inclusión al estudio. Todos acudieron secuencialmente en noviembre y diciembre del año 2001. Se purificó y se extrajo el ADN de cada muestra utilizando el sistema Wizard de Promega. Se realizó la amplificación por reacción en cadena de la polimerasa (PCR).
En el factor V se ha identificado la mutación A506 (FV Leiden). En el factor II la mutación G20210A, y en la MTHFR la mutación C677T. En el estudio del factor II se encontraron 5 individuos (Aa), con la mutación G20210A, con una prevalencia de 2,5 %. El Factor V Leiden se observó en 4 individuos heterocigotos (Aa), prevalencia de 2 %. La mutación C677T de la MTHFR se encontró en 85 individuos heterocigotos (Aa), y 27 individuos homocigotos (aa), con una prevalencia de 56 %.
Conclusiones: se han identificado las mutaciones más frecuentemente descriptas en los factores estudiados. La prevalencia del genotipo FII G20210A, FV Leiden y el gen codificante para la MTHFR, C677T, hallados en los individuos sanos estudiados, es similar a la hallada en la mayoría de los países latinos.
OBJETIVES. To identify one of the most frequent mutations and its prevalence, in the gene of Factor V, Protrombina, the MTHFR, in a population of donors of blood of Paraguay.
MATERIAL AND METHODS. Prospective observational study. For this work, blood samples of 200 donors of the Blood donation point have been taken from the Hospital of Clinics, that reunited the criteria from inclusion to the study. All sequentially went in November and December of year 2001. It was purified and the DNA of each sample was extracted using the Wizard system of Promega. The amplification by chain reaction was made of the polymerase (PCR). In factor V the A506 mutation has been identified (FV Leiden). In factor II mutation G20210A, and in the MTHFR mutation C677T. In the study of factor II, were 5 individuals (Aa), with mutation G20210A, a prevalence of 2.5 %. Factor V Leiden was observed in 4 heterocigotos individuals (Aa), prevalence of 2 %. Mutation C677T of the MTHFR was in 85 heterocigotos individuals (Aa), and 27 homocigotos individuals (aa), with a prevalence of 56 %.
CONCLUSIONS. The mutations have been identified more frequently you decipher in the studied factors. The prevalence of genotype FII G20210A, FV Leiden and the codificante gene for MTHFR, C677T, found in the studied healthy individuals, is similar to the found one in most of the Latin countries.
Las mutaciones en los dos primeros factores de la coagulación, es decir, en el factor II y en el V, son las causas más frecuentes de trombofilia hereditaria.
Cabe hacer notar, que este trabajo es cooperativo, entre el Banco de Sangre, el Departamento de Hematología de la Segunda Cátedra de Clínica Médica del Hospital de Clínicas de Asunción, y el Laboratorio de especialidades genéticas de Montevideo, Uruguay.
Antes de introducir al lector en el estudio, se hablará de generalidades sobre trombofilia, a modo de recuerdo. Se presentará además, un poco de historia, sobre los descubrimientos de los diferentes factores de riesgo genético, que se han ido haciendo a lo largo de los años.
Y finalmente se presentará la investigación propiamente dicha.
Los desórdenes de la hemostasia, en parte, pueden también estar genéticamente determinados, y cuando este es el caso, su influencia para el desarrollo de trombosis es profunda, ya que ese desorden está presente a lo largo de toda la vida.
Al igual que para la tendencia hemorrágica se acuñó el término hemofilia, se denomina trombofilia al estado de tendencia a padecer trombosis.
Se ha planteado que la trombofilia es una suma de factores endógenos que predisponen a la trombosis, la que se convierte en una trombosis manifiesta bajo la influencia de una exposición a factores exógenos 5.
La predisposición está dada por los factores endógenos (los defectos genéticos) o adquiridos, los cuales provocan en el individuo una disminución de la resistencia fisiológica hemostática para enfrentar diferentes alteraciones o fluctuaciones normales, producto de las interacciones con el ambiente.
La trombofilia hereditaria es definida como una proclividad genéticamente determinada, a la trombosis venosa, que ocurre en individuos, generalmente menores de 45 años, sin causa aparente y con riesgo de recurrencias. Los términos sinónimos son: estado de hipercoagulabilidad, estado pre o protrombótico, trombofilia primaria.
Se ha establecido que la trombofilia sigue el modelo conocido como herencia poligénica, es decir, que el rasgo clínico patológico requiere la presencia simultánea de mutaciones en varios genes5. Sin embargo, aún en la actualidad estos modelos genéticos no permiten explicar un buen número de casos de trombofilia.
Según el Comité conjunto de la Organización Mundial de la Salud (OMS) y la Sociedad Internacional de Hemostasia y Trombosis (ISTH), "la trombofilia hereditaria es una tendencia genéticamente determinada al tromboembolismo venoso. Tanto anomalías dominantes, en algunos casos, como combinaciones de defectos más leves, en otros, pueden expresarse clínicamente en edades tempranas, con recidivas frecuentes o con historia familiar de trombosis. Los rasgos leves pueden ser sólo revelados mediante investigación de laboratorio" 6.
Entre los defectos heredados de la hemostasia que causan trombofilia pueden citarse deficiencias de ciertas proteinas de la coagulación como la Antitrombina (AT), la proteina C (PC), la proteina S (PS). Y también ciertas mutaciones en los factores de la coagulación: Factor V Leiden, variante de la Protrombina G20210A, y mutaciones en la enzima MTHFR, entre otras.
Entendemos por mutación, cambios de nucleótidos que ocurren dentro de un gen, que puede afectar la función del gen en cuestión. La presencia de estas mutaciones en algunos factores proteicos de la hemostasia, pone en riesgo de trombosis a aquellos individuos que poseen una sola copia alterada, de las dos que tiene el gen: son los heterocigotos. Y pone en riesgo, más aún, a aquellos que poseen las dos copias mutadas; son los homocigotos.
En el año 1965 se describe el primer caso de deficiencia de AT, y por muchos años, ésta era la única causa identificada de trombofilia hereditaria 7.
A partir de la década de los años 80, ha habido una explosión de nuevos descubrimientos sobre el tema que nos ocupa: la trombofilia hereditaria. Las deficiencias de la PC y de la PS han sido las primeras identificadas, después de la de AT, como causas adicionales de trombofilia hereditaria 8.
Recordemos que la AT, la PC y la PS son proteínas inhibidoras fisiológicas de la coagulación.
Dahlback 9 en 1993 observó el fenómeno de resistencia a la proteína C activada, en un paciente con trombosis. Éste era un hombre de mediana edad con una historia personal y familiar de trombosis, cuyo test de Tiempo de Tromboplastina Parcial Activado (TTPA) no presentaba la prolongación espectada ante el agregado de Proteina C Activada (APC) exógena. El mecanismo era entonces desconocido. Un año más tarde era identificada una mutación puntual en el factor V, llamado Factor V Leiden 10,11. Corresponde a la sustitución especifica G-A en el nucleótido 1691 en el gen del factor V, mutación que determina la resistencia a la inactivación del factor V activado por el sistema de la proteína C (rAPC). Esta alteración en el factor V, es el factor de riesgo genético más frecuentemente encontrado en las trombosis venosas. La prevalencia del Factor V Leiden en los pacientes con trombosis es de 20 a 50 %, según diferentes series de estudio 12.
El sistema de la Proteina C (PC) antes referido, se basa en la acción de una enzima, la Proteina C activada (APC), que inhibe la generación de trombina por proteólisis de dos factores de la coagulación, el factor V activado (Va) y el Factor VIII activado (VIIIa). La PC circula en forma de un zimógeno vitamina K dependiente (como los factores II, VII, IX y X) que es activado por la trombina, fijada ésta, a un receptor presente en la superficie del endotelio vascular, la trombomodulina. De esta forma la protrombina pierde sus propiedades prohemostáticas para convertirse en activadora de un mecanismo antitrombótico. La interacción de la PC con sus substratos, los factores Va y VIIIa, requiere fijación a un fosfolípido, plaquetario o endotelial, y la presencia de un cofactor proteico, la Proteina S, que también es vitamina K dependiente.
Dahlback 9 en 1993, como ya se mencionó, y Bertina 11 en 1994, han hecho descubrimientos muy importantes: la resistencia a la Proteína C activada (rAPC) debido a una mutación puntual en el gen del Factor V (G1691A en el exon 10, llegando a Arg506Gln).
La APC es una serina proteasa que inhibe la actividad de la cascada de la coagulación inactivando los factores de la coagulación, Va y VIIIa. La PCA es altamente específica en su acción y no tiene efecto sobre las formas circulantes inactivas de los factores V y VIII. Como decíamos antes, la actividad anticoagulante de la PCA es potenciada por un cofactor, la PS. Se sabe también que el factor V intacto potencia la actividad anticoagulante de la PS. Y la hipótesis es que las dos proteinas: el factor V y la PS funcionan como cofactores sinérgicos de la APC. La activación del factor V por la trombina, está asociada con la pérdida de su actividad anticoagulante. Concomitantemente la actividad procoagulante es ganada porque el factor Va es un cofactor para el factor X activado (Xa) en la activación de la protrombina. La doble función del factor V se suma a la lista de mecanismos ingeniosos que bajo condiciones fisiológicas balancean las fuerzas pro y anticoagulantes 8.
La mutación en el factor V está asociada a riesgo de trombosis venosa, entre 6 a 8 veces más que los individuos que no tienen tal mutación 12,13,14,15.
La importancia de los factores de riesgo genéticos en la producción de trombosis, ha aumentado con la puesta en evidencia del descubrimiento de Bertina y colaboradores. Ellos encontraron un polimorfismo genético en el factor II, la variante de la Protrombina G20210A 16,17.
La variante alélica protrombina G20210A 16,17 es una mutación en el gen de la protrombina, asociada a un aumento de la concentración plasmática de protrombina y que confiere un mayor riesgo de trombosis venosa a los individuos portadores. Esta mutación tiene una frecuencia de 6 % en pacientes con un primer episodio de trombosis venosa. La frecuencia en la población blanca general, sin episodio de trombosis, es de aproximadamente 2 % 15.
Y por último, la mutación en el gen de la enzima metiltetrahidrofolato reductasa (MTHFR) responsable del aumento de la homocisteína en sangre, en los pacientes homocigotos y en los heterocigotos, es también causa de trombofilia hereditaria 4, como ya se dijo antes.
A diferencia de las causas de hipercoagulabilidad hereditaria: déficit de AT, PC, PS y Factor V Leiden, en las cuales la alteración se halla en un solo gen, en la hiperhomocisteinemia hereditaria los defectos pueden encontrarse en varios genes que codifican las diferentes enzimas comprometidas en el metabolismo del aminoácido.
La hiperhomocisteinemia de causa hereditaria se debe a mutaciones en los genes de las enzimas cistationina b sintasa y/o metilen tetrahidrofolato reductasa (MTHR), que conlleva a alteraciones del metabolismo de la metionina con la acumulación de la homocisteína en sangre.
La causa más frecuente de una hiperhomocisteinemia severa, con niveles totales de homocisteína superiores a 100 mmol/L, es la deficiencia homocigótica de la cistationina b sintasa 18. La moderada (25-100 mmol/L) o suave (16-24 mmol/L) son típicas de individuos heterocigotos y de la variante termolábil de la MTHFR. Esta última forma es la más frecuente. El estado heterocigoto para la cistationina b sintasa es poco frecuente 18.
En ambos casos de mutaciones, ya sea de la cistationina b sintasa o de la MTHFR, los individuos afectados sufren de enfermedades vasculares prematuras y tromboembolismo 18. El mecanismo por el cual estas alteraciones contribuyen a la aterogénesis y a la trombogénesis es parcialmente comprendido. Se ha demostrado que se produce una descamación del endotelio vascular, activación del factor V, interferencia en la activación de la PC y de la expresión de la trombomodulina, inhibición del activador tisular del plasminógeno, interferencia en la producción de óxido nítrico y prostaciclina 18, entre otros.
En la hiperhomocisteinemia las manifestaciones de tromboembolismo venoso no difieren del resto de los síndromes trombofílicos. La manifestación clínica más frecuente es la trombosis venosa profunda con la complicación de un tromboembolismo pulmonar o sin él.
Ya en el año 1969 19 se habló de la presencia de trombosis arterial y aterosclerosis en la necropsia de 2 niños con hiperhomocisteinemia, y se relacionó por primera vez la concentración elevada de homocisteína plasmática con la enfermedad vascular. Posteriormente otras investigaciones confirmaron la hipótesis y establecieron esta anomalía como factor de riesgo independiente, para padecer trombosis 20.
La homocisteína es un aminoácido sulfurado formado durante el metabolismo de la metionina. Es un aminoácido esencial derivado de las proteínas de la dieta.
La hiperhomocisteinemia, es decir su elevación en plasma, se produce más frecuentemente, como se dijo antes, por defectos genéticos de la enzima 5,10-metilentetrahidrofolato reductasa, que permite la reconversión de homocisteína a metionina, mediante la remetilación, y con menor frecuencia por carencias nutricionales de cofactores vitamínicos esenciales: vitaminas B6, B12 y ácido fólico 21.
Numerosos estudios consideran a la hiperhomocisteinemia como factor de riesgo de enfermedad oclusiva arterial y venosa 21. En efecto, Fernández-Miranda C y col. 21 en un estudio realizado en 202 enfermos coronarios demostraron que el 26% de ellos presentaba hiperhomocisteinemia, aunque otro trabajo 20 mostró que su intensidad no predijo fiablemente la gravedad de las lesiones coronarias encontradas.
Generalmente, en los portadores de hiperhomocisteinemia los síntomas no aparecen sino en la tercera o cuarta décadas de la vida, época en que se desarrolla de forma prematura la enfermedad arterial coronaria, así como trombosis recurrentes del sistema arterial y venoso.
Algunos investigadores 21 afirman que la hiperhomocisteinemia se debe a disfunción endotelial y anormal función plaquetaria. Dicen además, que el aporte de complejo B y ácido fólico puede normalizar las concentraciones de homocisteína, habitualmente a las 4-6 semanas de iniciada la terapia, aunque no se conoce el impacto de este tratamiento sobre la morbimortalidad cardiovascular. Estas incertidumbres obligan, hoy día, a no aconsejar con carácter general la determinación sistemática de la concentración plasmática de homocisteína, salvo en pacientes con aterosclerosis prematura no explicable por presencia de factores de riesgo cardiovascular conocidos.
Sin embargo, se sabe muy bien que las mutaciones en el gen de la MTHFR son una causa de Homocistinuria 17. El polimorfismo de la enzima (en el cual una sustitución del aminoácido valina por alanina), trae como consecuencia una disminución en la actividad enzimática de la MTHFR, y por ende una elevación de la homocisteina en sangre. En algunos estudios 24, este polimorfismo está asociado a una franca disminución de la densidad ósea o a un incremento del riesgo de fractura ósea. En estos casos, según las investigaciones, esta sustitución de la valina en el polimorfismo de la enzima, está asociado a niveles bajos de ácido fólico en sangre 2.
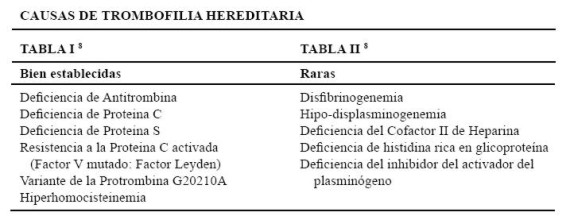
En la tabla I figura la lista de las causas más frecuentes y bien establecidas de trombofilia hereditaria. En la tabla II las causas más raras, según De Stefano 8.
La investigación la hemos desarrollado en donantes del Banco de Sangre del Hospital de Clínicas, en el lapso de los meses de noviembre y diciembre del año 2001. Se han involucrado en este estudio el Departamento de Hematología de la II Cátedra de Clínica Médica, el Banco de Sangre del Hospital de Clínicas de Asunción y el laboratorio de especialidades génicas de Montevideo, Uruguay.
Han ingresado al estudio todos los donantes de sangre que han reunido los criterios de inclusión.
Los Criterios de inclusión son los de selección del donante de sangre que figura en el Anexo 1. Y conforme a los criterios para la protección del donante y del receptor. Hay así mismo un apartado de información al donante.
Los criterios de exclusión, figuran igualmente en el Anexo 1, y son conforme a normas internacionales.
Para estimar el tamaño de muestra, se tomó al Factor V Leiden, cuya proporción esperada en orden a los estudios sobre todo del norte de Europa es de aproximadamente 7 %. Y así para un índice de confianza de 95 %, y una amplitud total del intervalo de confianza (W) de 0,10 el número total de donantes de sangre era de 139 individuos. Nosotros hemos tomado material de 200 donantes.
Las muestras fueron transportadas desde Asunción, vía aérea y conforme a normas reglamentarias de transporte para este tipo de material hasta el laboratorio de especialidades génicas de Montevideo, Uruguay. Se utilizó para la investigación, 100µl de sangre del material extraido para el efecto.
Se purificó el ADN contenido en las muestras entre 2 y 14 meses después de obtenida la sangre. Antes de procesarla, se conservó la misma entre 2 y 8 °C.
Como controles positivos se utilizó ADN proveniente de individuos homocigotos mutados para cada factor, es decir, mutados para el factor II, para el factor V y para la enzima MTHFR.
El método se basa, en su forma más simple en la realización de tres reacciones sucesivas llevadas a cabo a distintas temperaturas. Estas reacciones se repiten cíclicamente entre veinte y cuarenta veces.
La muestra se calienta, en el primer paso, hasta lograr la separación de las dos cadenas que constituyen el ADN, hecho que se conoce como "desnaturalización".
En el segundo paso, la temperatura se reduce para permitir el "apareamiento" de cada una de dos cadenas cortas de nucleótidos (oligonucleótidos) con cada una de las hebras separadas del ADN molde. Se trata de segmentos de ADN de cadena simple, sintetizados en el laboratorio y diseñados de manera tal que permiten definir los límites del tramo de ADN que se desea replicar. Para que se pueda producir el apareamiento, cada uno de estos oligonucleótidos, a los que se denomina "iniciadores" o primers, debe ser complementario del tramo al que tienen que unirse en las cadenas separadas del ADN molde.
En tercer lugar, una enzima ADN polimerasa extiende los primers, en el espacio comprendido entre ambos, sintetizando las secuencias complementarias de las hebras del ADN molde. Para ello, la ADN polimerasa usa desoxidonucleósidos trifosfatos (DNTPs) agregados a la mezcla de reacción. La temperatura a la que se realiza el tercer paso es la temperatura en que la ADN polimerasa posee mayor actividad. Al cabo del primer ciclo de tres reacciones (desnaturalización, apareamiento, extensión) el tramo de ADN elegido se ha duplicado y el doble de su cantidad original se encuentra disponible para ser nuevamente replicado en un segundo ciclo. El resultado de la aplicación de numerosos ciclos sucesivos da lugar a la amplificación geométrica del segmento de ADN delimitado por los primers.
Una vez finalizada la reacción se logra sintetizar, en pocas horas, un gran numero de copias de un fragmento génico con un alto grado de pureza.
La determinación de Factor V Leiden, la mutación de la MTHFR y el polimorfismo G20210A de la variante de la protrombina se determinó por PCR, seguido por las apropiadas enzimas de restricción.
El programa utilizado ha sido el Microsof Excel XP.
Para las variables cualitativas se usó la Frecuencia Relativa (frecuencias génicas y fenotípicas). La prevalencia puntual se calculó con la siguiente fórmula: 
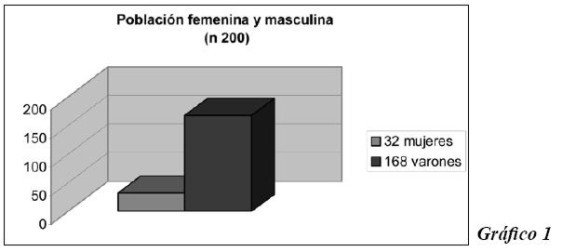
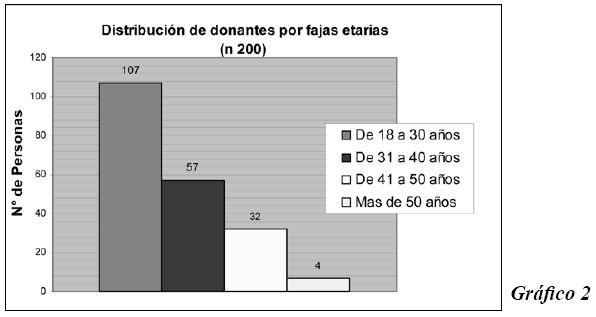
Primeramente se hace una descripción de la población estudiada en lo que corresponde a distribución por sexo, edades y procedencia. En el Gráfico 1, se observa que de 200 individuos, 32 eran mujeres y 168 varones. El Gráfico 2 muestra que más del 50 % de personas eran jóvenes de entre 18 y 30 años.
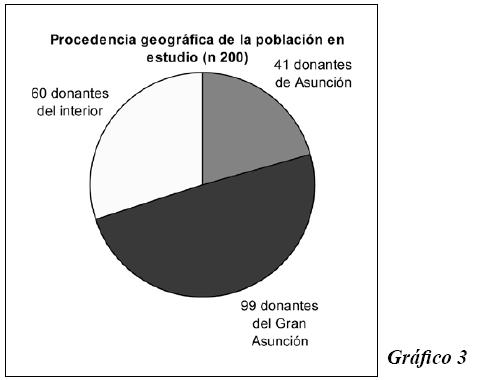
El Gráfico 3 ilustra la procedencia geográfica de los individuos estudiados. La mayoría procedía de zonas aledañas a la capital, es decir de las ciudades cercanas como Lambaré, San Lorenzo, Itaguá, Luque, Limpio, Mariano Roque Alonso, Capiatá, Fernando de la Mora, entre otras. (Gráfico 3)
Se presentan los gráficos de cada factor, por separado.
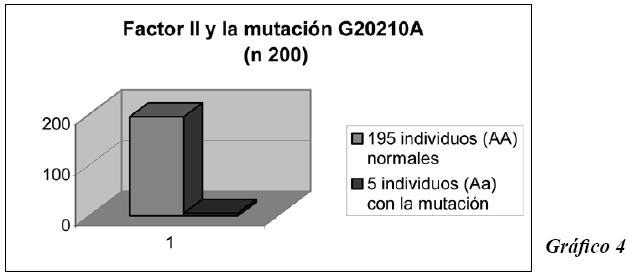
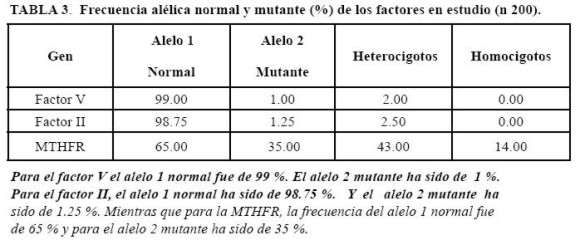
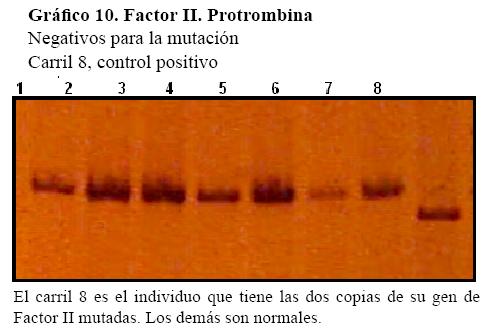
En el Factor II se observaron 195 individuos (AA) que son normales, con una frecuencia genotípica de 0.98 y 5 individuos (Aa), heterocigotos para la mutación G20210A, con una frecuencia de 0.02. Gráfico 4.
Los 5 individuos (Aa), mutados, son heterocigotos para la mutación G20210A.
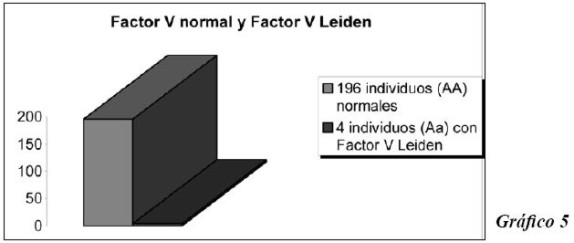
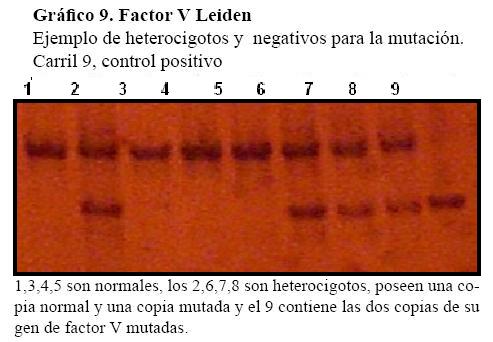
En el factor V se observaron 196 individuos homocigotos normales (AA) con una frecuencia genotípica de 0.98, y 4 individuos (Aa), heterocigotos para la mutación Factor V Leiden, con 0.02 de frecuencia. Gráfico 5.
Los 4 individuos (Aa) mutados, son heterocigotos para la mutaciòn F V Leiden
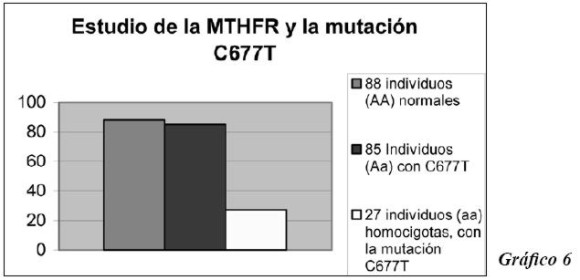
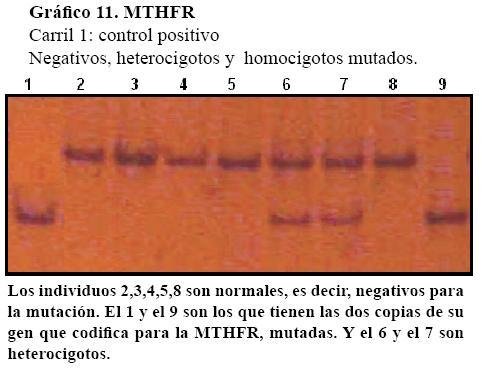
Finalmente, en el gen que codifica para la enzima MTHFR se encontraron 88 individuos normales (AA), cuya frecuencia es de 0.44; 85 individuos (Aa), heterocigotos para la mutación C677T con 0.43 de frecuencia genotípica; y 27 (aa) homocigotos para la alteración señalada, con una frecuencia genotípica de 0.14. Gráfico 6.
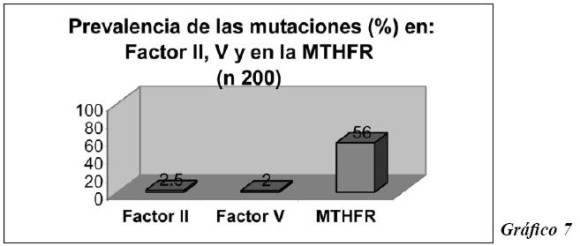
Y en el gráfico 7 se ilustra la prevalencia de las mutaciones de los factores estudiados que son: 2.5% para la C677T, 2.0 % para el Factor V Leiden y 56.0 % para la enzima MTHFR.(Gráfico 7)
Para el factor II, el alelo 1 normal ha sido de 98.75 %. Y el alelo 2 mutante ha sido de 1.25 %. Mientras que para la MTHFR, la frecuencia del alelo 1 normal fue de 65 % y para el alelo 2 mutante ha sido de 35 %.
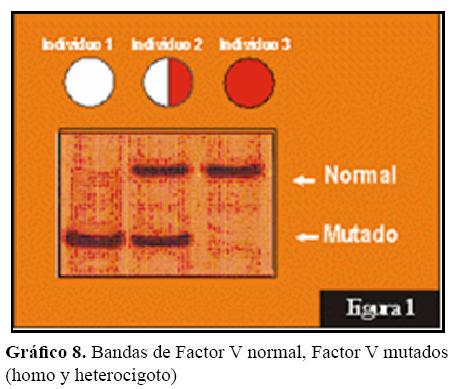
En las figuras que siguen se observan algunas muestras de bandas de factores normales y mutados. (Gráfico 9, Gráfico 10 y Gráfico 11)
El gráfico 8 ilustra bandas de individuos con Factor V normal, con factor V mutado, es decir, con Factor V Leiden, homo y heterocigoto. La banda superior de ADN corresponde a la copia del gen de factor V normal y la inferior a la copia mutada.
Las frecuencias génicas y fenotípicas halladas en este trabajo, están dentro del margen esperado, en comparación con las estudiadas por algunos investigadores. Es decir, que los resultados obtenidos en este estudio muestran que la frecuencia génica obtenida para las mutaciones en estudio coincide con la esperada en una población en equilibrio de Hardy-Weinberg.
En lo que respecta al Factor V Leiden se han encontrado prevalencias que no difieren tan grandemente de la nuestra. En el estudio de la Dra. Guggiari 1, la frecuencia es de1, 66 %. Y en lo que respecta específicamente a las poblaciones vecinas e indígenas tenemos, Venezuela (Valencia) 1,6%; Costa Rica 2,0%; Brasil (San Pablo) 2% y Chile (Santiago) 3,8% 23,24,25. La población capitalina de la República Argentina tiene, sin embargo, una prevalencia mayor 5,1%; Las diferencias son mayores en otras regiones más alejadas: en Chipre 12%; en el Reino Unido: 3 a 8%; en americanos caucásicos, 5.2%. En tanto que, en países latinos como Italia y España la frecuencia es de 2% 25,26,27, igual a la nuestra.
El conjunto de deficiencias de AT, de PC y de PS sólo llega a causar menos del 15 % de trombosis juvenil o recurrente en una población seleccionada, y menos del 10 % en una población de casos no seleccionados 3.
Por el contrario, el factor V Leiden es mucho más prevalente. Este descubrimiento, dice P Mannuci 27, ha sido confirmado y citado más de 2000 veces en la literatura médica mundial. El estado de heterocigoto para el factor V Arg506GL, conocido más frecuentemente como Factor V Leiden, está presente en aproximadamente el 20 % de pacientes con un primer episodio de trombosis. Y lo que es aún más importante, esta mutación está presente en el 4 % de la población blanca en general, lo cual pone en riesgo a los portadores.
Otra observación que merece la pena subrayar con respecto al Factor V Leiden son los hallazgos del Copenhagen City Heart Study 29 que dice que si bien es cierto que el Factor V Leiden se asocia a Trombosis venosa, la asociación con trombosis arterial no es posible afirmar; y esto en lo que respecta a infarto agudo de miocardio, cardiopatía y stroke isquémicos 27.
Adicionando al Factor V Leiden y a la Protrombina G20210A, las raras deficiencias de las proteínas inhibidoras fisiológicas de la coagulación, como la AT, la PC, la PS, la proporción de trombosis venosas atribuibles a los factores genéticos hereditarios es de 25 a 30 %. Esta proporción es, al menos, tan grande e importante 27, como las trombosis atribuibles a los bien establecidos factores de riesgo adquiridos, tales como cirugía, trauma, puerperio, inmovilización prolongada en cama (más de 7 días), embarazo, toma de anticonceptivos orales, neoplasias y terapia hormonal de reemplazo.
Con estos factores de riesgo genético de trombosis venosas con tal peso y prevalencia en la población general, la primera cuestión que se nos plantea es de si, esos factores deberían estudiarse en los individuos sanos sin historia familiar ni personal de trombosis, cuando ellos se exponen a factores adquiridos que pueden interactuar con factores genéticos incrementando el riesgo de trombosis.
La prevalencia de la mutación en la enzima MTHFR es muy alta en nuestro estudio, de 56 %. Y podrían estar apareciendo casos de trombofilia hereditaria por mutación en la enzima MTHFR, que no se diagnostican. Es preciso recordar que la trombosis es una enfermedad multifactorial. Y si a esa situación de riesgo genético se asocian otros factores de riesgo del medio ambiente o circunstancial, las probabilidades de desarrollar la enfermedad es bastante elevada. Es verdad, que los estudios de mutaciones de los factores causantes de trombofilia hereditaria no se realizan ordinariamente en nuestro medio, lo cual dificulta el verdadero diagnóstico. Sin embargo, aquí vale aquel dicho de que, la ausencia de evidencias (y me refiero a los estudios de las mutaciones) no evidencia la ausencia.
En nuestro estudio, el rango de edades en el que aparece el 100 % de la mutación en el factor II, Protrombina G20210A, es el de 18 a 30 años.
Es la misma faja etaria afectada en su mayoría, para el Factor V Leiden.
Sin embargo, la mutación en la enzima MTHFR, la C677T se encuentra en todas las fajas etarias. Estas van de los 18 a los 73 años. Y precisamente, el paciente de 73 años que es el de más edad en el estudio es portador de la mutación.
La cuestión que se nos plantea es el por qué, de la presencia de las mutaciones en los factores II y V en los más jóvenes. Sólo un portador del Factor V Leiden tenía 41 años. El resto de la población afectada por estas dos mutaciones era en su mayoría perteneciente al rango de edades entre 18 y 30 años. No es posible que solo en este periodo de tiempo se hayan establecido las mutaciones. No. ¿Qué pasó entonces con los portadores que ahora tendrían más de 40 años? ¿Es casualidad y azar el que solo se vean estas alteraciones en la mayoría de los menores de 30 años?
Motivo de investigación sería determinar el riesgo relativo para el desarrollo de trombosis, en una cohorte de seguimiento de los individuos portadores.
Cabe señalar también, que hay otras causas de trombofilia, descubiertas en la última década. Estudios serios plantean el tema. Así, se asocian altos niveles del factor VIII 30, del factor XI 31, del factor IX y TAFI 29 con incremento del riesgo de trombosis venosas.
Tampoco sabemos si los donantes estudiados aquí, son portadores de otros defectos hereditarios que coexisten y triplican o cuadriplican el riesgo de trombosis, más aún si se suman los factores de riesgo adquiridos.
Y para tener datos seguros y precisos sería interesante poder hacer un screening como los que plantea en su artículo De Merloose 32. Y todo esto, respondiendo a las preguntas: ¿Qué estudios, qué tests? ¿A cuáles pacientes? ¿Al que es familiar de alguien con trombosis a repetición, o porque es portador de algún otro estigma familiar o personal? ¿A quién enviaremos a los pacientes para realizar los estudios? (ya que depende de la garantía que puedan dar los resultados: formación técnica y científica, equipos confiables, experiencia, excelencia en calidad, etc.). ¿Cuándo realizar los estudios? ¿Y por qué realizar esos estudios?
Y finalizando, en nuestro medio y a la luz de los conocimientos actuales, todavía hay mucho que investigar en orden a la trombofilia hereditaria. Y los caminos no son tan llanos.
2. Lens D, Otero AM, Cotic G, Henry S, Díaz A, Attarian D, Agorio C, Piern S. Diagnóstico molecular de factores protrombóticos: primeros casos de factor V Leiden y protrombina 20210ª en Uruguay. Rev Med Urug 2000; 16: 39-44. [ Links ]
3. Annichino-Bizzachi J. Resistencia a Proteína C ativada. Libro del Simposio Internacional del Grupo CLATH. Rio de Janeiro 1998: 57-62. [ Links ]
4. Lane DA, Grant PJ. Role of hemostatic gene polymorfhisms in venous and arterial thrombotic desease. Blood 2000; 95: 1517-32. [ Links ]
5. Souto JC. Trombofilia: enfermedad poligénica. Rev Iberoam Tromb Hemostasia 1997; 10: 114-32. [ Links ]
6. Lane DA, Mannucci PM, Bauer KA, Bertina R, Bochkov NP, Bpulyjenkov V. Inherited thrombophilia. part. 1. Thromb Haemost 1996; 76: 651-2. [ Links ]
7. Lane DA, Mannucci PM, Bauer KA, Bertina R, Bochkov NP, Bpulyjenkov V. Inherited thrombophilia. part. 2. Thromb Haemost 1996; 76: 824-34. [ Links ]
8. De Stefano V, Finazza G, Mannucci PM. Inherited Thrombophilia: Pathogenesis, Clinical Syndromes, and Management. Blood 1996; 87: 3531-44. [ Links ]
9. Dahlback B, Carlsson M, Svensson PJ: Familial thrombophilia dueto a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: Prediction of a cofactor to activated protein C. Proc Natl Acad Sci 1993, USA 90:1004. [ Links ]
10. Bertina RM, Koeleman RP, Koster T et al. Mutation in blood coagulation factor V associated with resistance to activated Proteina C. Nature 1994; 369: 64-67. [ Links ]
11. Svensson PJ, Dahlback B: Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med 1994; 330: 517. [ Links ]
12. Simioni P., Prandoni P., Lensing A. W.A., Scudeller A., Sardella C., Prins M. H., Villalta S., Dazzi F., Girolami. The Risk of Recurrent Venous Thromboembolism in Patients with an Arg506 Gln Mutation in the Gene for Factor V (Factor V Leiden). N Engl J Med 1997; 336:399-403. [ Links ]
13. Gerhardt A,Scharf RE, Beckmann M W, Struve S, Bender HG, Pillny M, Sandmann W, Zotz RB. Prothrombin and Factor V Mutations in Women with a History of Thrombosis during Pregnancy and the Puerperium. N Engl J Med 2000; 342:374-380. [ Links ]
14. Simione P, Tormene D, Prandone P, Zerbinate P, Gavasso S, Cefalo P, Girolami A. Incidence of venous thromboembolism in asymptomatic family members who are carriers of factor V Leiden: a prospective cohort study. Blood 2002; 99: 1938-42. [ Links ]
15. De Stefano V, Martinelli I, Mannucci PM, Paciaroni K, Chiusolo P, Casorelli I, Rossi E, Leone G. The Risk of Recurrent Deep Venous Thrombosis among Heterozygous Carriers of Both Factor V Leiden and the G20210A Prothrombin Mutation. N Engl J Med 1999; 341:801-806. [ Links ]
16. Mandel H, Brenner B., Berant M, Rosenberg N, Lanir N, Jakobs C, Fowler B, Seligsohn U. Coexistence of Hereditary Homocystinuria and Factor V Leiden Effect on Thrombosis. N Engl J Med 1996; 334:763-768. [ Links ]
17. Almeida DP. Trombofilia primaria, características de una enfermedad poligénica. Rev Cubana Angiol y Cir Vasc 2000; 1: 14-54. [ Links ]
18. McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969; 56:111-28. [ Links ]
19. Falcon CR, Cattaneo M, Panzeri D, Martinelli I, Mannucci PM. High prevalence of hyperhomocyst(e)inemia in patients with juvenile venous thrombosis. Arterioscler Thromb 1994; 14: 1080. [ Links ]
20. Fernández-Miranda C, Aranda JL, Gómez GP, Díaz-Rubio P, Estenoz J, Gómez de la Cámara A. La hiperhomocisteinemia es frecuente en pacientes con enfermedad coronaria. Estudio de 202 enfermos. Med Clin 1999; 113:407-10. [ Links ]
21. Rodríguez JF, Escobales N, Cruz D, Banch H, Rivera C, Altieri PI. Concentraciones totales de homocisteína plasmática en pacientes puertorriqueños con cardiopatía isquémica. Rev Esp Cardiol 2001; 54:1411-6. [ Links ]
22. Van den Berg M, Boers GH, Franken DG, Blom HJ, Van Kamp GJ, Jakobs C, et al.. Hyperhomocysteinaemia and endothelial dysfunction in young patients with peripheral arterial occlusive disease. Eur J Clin Invest 1995; 25: 176-81. [ Links ]
23. Abrahamsen B, Madsen JS, Tofteng CL, et al. A common methylenetetrahydrofolate reductase (C677T) polymorphism is associated with low bone mineral density and increased fracture incidence after menopause: longitudinal data from the Danish osteoporosis prevention study. J Bone Miner Res 2003;18:723-729. [ Links ]
24. Boushey CJ, Bereford SA, Omenn GS, Motulsky AG.. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease: probable benefits of increasing folic acid intakes. JAMA 1995; 274:1049-57. [ Links ]
25. Ramacciotti E, Wolosker N, Puech-Leao P, Zeratti EA, Gusson PR, del Giglio A, Franco RF. Prevalence of factor V Leiden, FII G20210A, FXIII Val34Leu and MTHFR C677T polymorphisms in cancer patients with and without venous thrombosis. Thromb Res 2003; 109: 171-4. [ Links ]
26. Arruda VR, Annichino Bizzacchi JM, Goncalves MS, Costa FF. Prevalence of the prothrombin gene variant (nt 20210A) in venous thrombosis and arterial disease. Thromb Haemost 1997; 78: 1430-3. [ Links ]
27. Mannucci P. Genetic hipercoagulability: prevention suggests testing family members. Blood 2001; 98: 21-2. [ Links ]
28. Klaus Juul, Anne Tybjærg-Hansen, Rolf Steffensen, Steen Kofoed, Gorm Jensen, and Børge Grønne Nordestgaard. Factor V Leiden: The Copenhagen City Heart Study and 2 meta-analyses. Blood 2002, 100: 3-10. [ Links ]
29. Bertina RM. Genetic approach to thrombophilia. Thromb Haemost 2001; 86: 92-103. [ Links ]
30. Kyrle PA, Minar E, Hirschl M, Bialonczyk C, Stain M, Schneider B, Weltermann A., Speiser W., Lechner K., Eichinger S. High Plasma Levels of Factor VIII and the Risk of Recurrent Venous Thromboembolism. N Engl J Med 2000; 343:457-462. [ Links ]
31. Meijers JC.M, Tekelenburg W. LH, Bouma BN., Bertina RM., Rosendaal FR. High Levels of Coagulation Factor XI as a Risk Factor for Venous Thrombosis. N Engl J Med 2000; 342:696-701. [ Links ]
32. De Moerloose P, Bounameaux HR, Mannucci PM. Screening test for thrombophilic patients: which tests, for which patient, by whom, when, and why? Semin Thromb Hemost 1998; 24: 321-7. [ Links ]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}