









Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Ciencias Médicas (Asunción)
versión impresa ISSN 1816-8949
An. Fac. Cienc. Méd. (Asunción) v.38 n.1-2 Asunción abr. 2005
ARTICULO DE INTERES
Manejo multidisciplinario y diagnóstico prenatal de la distrofia miotónica del adulto en el embarazo.
Multidisciplinary management and prenatal diagnosis of myotonic distrophy adult from in pregnancy.
Miguel Ruoti Cosp1-2, Ernesto Fabre González1, Lia Ornat Clemente1, Rafael González de Agüero Laborda1, María Jesús Barco Marcellán1, Mercedes Sobreviela Laserrada1, Oros Lopez Daniel1, Cristina Torrijo1.
1) Sección de Ecografía y Diagnóstico Prenatal. Departamento de Obstetricia y Ginecología. Hospital Clínico Universitario Lozano Blesa. Zaragoza. España. 2) Departamento de Medicina Perinatal. Cátedra de Ginecología y Obstetricia. Centro Materno Infantil. Facultad de Ciencias Médicas. U.N.A.
RESUMEN
La distrofia miotónica del adulto (DMA), es una rara afección neuroendócrina degenerativa, autosómica dominante, caracterizada principalmente por miotonía y debilidad muscular. Su infrecuente asociación con el embarazo por el hipogonadismo que generalmente acompaña, agravada su cuadro clínico además de complicar el curso gestacional por la presencia de polihidrmanios, partos prematuros, hemorragias en el post parto, entre otras. La diversidad de órganos y sistemas que afectan amerita la intervención de otras especialidades a fin de obtener un buen resultado perinatal. Presentamos el caso clínico de una gestante con antecedentes familiares y clínica actual de DMA y distrofia miotónica congénita diagnosticada prenatalmente, en el que se recurrió a la formación de un equipo multidisciplinario tanto para su manejo clínico, obstétrico, del diagnóstico prenatal y desenlace final.
Palabras claves: distrofia miotónica del adulto, embarazo, manejo multidisciplinario, diagnóstico prenatal.
SUMMARY
Myotonic Dystrophy adult form (MDA) is a rare neuroendocrine degenerative disease of genetic origin with autosomal dominant inheritance. Its main characteristics are myotonic dystrophy and muscular weakness. MDA is infrequently associated with pregnancy, since patients often have hypogonadism. Pregnancy is often aggravated with the presence of hydramnios and other common complications are; premature birth and post partum haemorrhages. This disease affects a number of different organs and systems, for that reason it deserves the formation of a multidisciplinary team to attend this group of patients. We present the case of a pregnant woman with a positive family history and the clinical characteristics of MDA, with the prenatal diagnosis and the suspicion of congenital Myotonic Dystrophy in her offspring. We created a multidisciplinary team for the clinical and obstetric management of the mother and her child to try and improve the final outcome.
Key words: Myotonic Distrophy, pregnancy, multidisciplinary management, prenatal diagnosis.
INTRODUCCION
La distrofia miotónica es una rara enfermedad neuroendócrina degenerativa. Descrita por Steinert en 1990, es la distrofia muscular del adulto (DMA) más común, cuya incidencia oscila en 13,5 por cada 100.000 nacidos vivos1.
Se trasmite siguiendo un patrón autosómico dominante con penetrabilidad casi completa y grado de expresividad variable, siendo el gen responsable (miotonina protein kinansa) localizado en la región q13.3 del brazo largo del cromosoma 19 y caracterizado por el aumento en el número de tripletes CTG (citosina-timina-guanina) en 3´ de su región no codificante2. Afecta de igual manera a ambos sexos1.
La gravedad clínica se relaciona estrechamente con el tamaño de la secuencia de copias de estos tripletes, cuanto mayor es su número mayor gravedad. En la población normal se encuentran entre 5-27 copias, portadores sanos pero transmisores de la enfermedad con 27-50 y manifiestan síntomas clínicos a partir de las 50 copias, reportándose varios miles en la forma mas grave, la distrofia miotónica congénita (DMC) 3.
La DMA se presenta en general, luego de la pubertad con miotonía, expresada por la contracción sostenida y prolongada de los músculos tras una percusión o estimulación eléctrica, en especial en manos y lengua, por lo que es típico su diagnóstico al estrechar la mano del afecto y comprobar que no puede soltarla luego1.
Con posterioridad se instaura un cuadro de atrofia y debilidad muscular distal en los miembros, además de la musculatura palatina, faríngea y lingual produciendo disartria, voz nasal y dificultad para la deglución respectivamente. Afecta también el elevador de los párpados provocando ptosis palpebral y el masetero, causando adelgazamiento de la mitad inferior de la cara que junto a la calvicie, habitualmente frontal en los hombres y parcial en las mujeres, determinan una facie típica de cara enjuta 4.
Con la evolución de la enfermedad también se puede producir afección de la musculatura proximal y del tronco confinándo a una silla de ruedas luego de 15 a 20 años de inicio de la enfermedad, así como debilidad del diafragma y de los músculos intercostales llevando a un cuadro de insuficiencia respiratoria con disminución de la ventilación que se traduce en bronquiectasias e infecciones bronquiales5.
Otras de las manifestaciones como trastornos cardiacos de conducción (bloqueos cardiacos de grado variable), cataratas subcapsulares posteriores, deterioro intelectual, hipersomnia, atrofia gonadal, disfunciones tiroideas, resistencia a la insulina o alteraciones en el transito intestinal, pueden estar presentes6, así como complicaciones anestésicas e hipertermia maligna1.
Su diagnóstico es fundamentalmente clínico pero algunas veces es necesario recurrir al electromiograma o bien a la biopsia muscular4.
Si bien es poco frecuente su asociación con el embarazo por el hipogonodasimo que puede acompañar al cuadro, la gestación causa deterioro del estado clínico con exacerbación de la sintomatología en especial la debilidad y miotonía, volviendo a mejorar en el puerperio7, así como puede conducir a complicaciones en el embarazo (aborto, polihidrmanios, parto prematuro, hemorragias puerperales, entre otras) 8.
El diagnóstico prenatal, obligado al ser una entidad en el que el 50% de la descendencia de los enfermos resultará afecto, se puede realizar mediante la obtención de células fetales por amniocentesis o biopsia corial, determinando el número de copias de tripletes9,10 y proporcionar un consejo genético a la pareja ante la posibilidad de solicitar la interrupción voluntaria del embarazo, en virtud de la gravedad de la DMC11, cuando la legislación lo permita.
La diversidad de órganos y sistemas que afectan la DMA y en especial cuando se produce el embarazo, amerita la intervención de otras especialidades a fin de obtener un buen resultado perinatal. Presentamos el caso clínico de una gestante con antecedentes familiares y clínica actual de DMA con DMC diagnosticada prenatalmente, en el que se recurrió a la formación de un equipo multidisciplinario tanto para su manejo clínico, obstétrico, del diagnóstico prenatal y desenlace final.
CASO CLÍNICO
Primigesta de 24 años de edad con DMA desde hace 8 años, con antecedentes familiares expuestos en la figura 1. Clínicamente presenta bradipsiquia, hipersomnolencia, miopatía en manos y mandíbula, debilidad en manos, dificultad para iniciar la de ambulación, disfagia y calvice parcial. El examen físico revela pares craneales normales salvo cierta atrofia de músculos maseteros, extremidades con disminución de la fuerza distal para la dorsifelxión de ambos pies (4+/5), marcada dificultad para caminar sobre los talones, fenómeno miotónico en manos y reflejos rotulianos abolidos distalmente.
A la 16ª semana de gestación se realiza amniocentesis, cariotipo 46 XX, con rendimiento de la extracción de ADN de los amniocitos escaso, sin posibilidad de amplificación de PCR informativa (una única banda dentro del rango de la normalidad). Se informa a la pareja de los resultados obtenidos quienes deciden continuar con el embarazo.
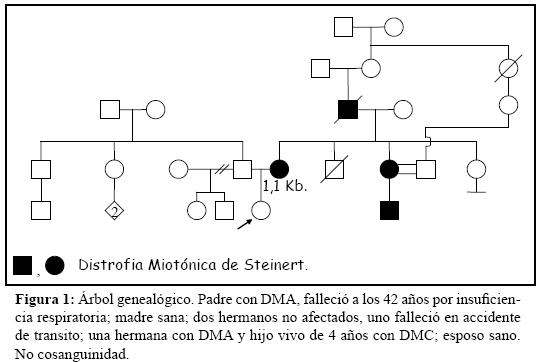
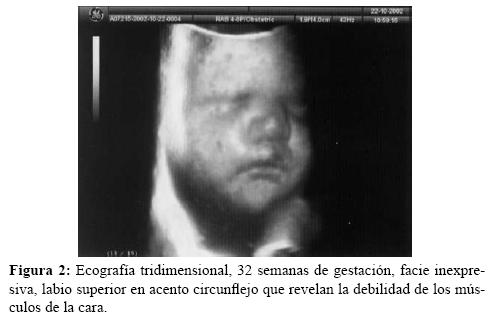
A la 20ª semana no se objetivan anomalías estructurales en la ecografía de diagnóstico prenatal. 32ª semana en visita rutinaria, la madre refiere disminución subjetiva de los movimientos fetales. La ecografía revela ILA de 28 (polihidramnios), escasa movilidad fetal, facie inexpresiva con la comisura de los labios superiores hacia abajo (en acento circunflejo o boca en triángulo) (figura 2), extremidades inferiores en hiperextensión (figura 3), pie equino varo con desviación cubital del primer dedo de ambos pies y CIR tipo II con flujometría Doppler normal. En controles ecográficos posteriores hasta la finalización de la gestación se confirman estos hallazgos, acompañados en períodos de bruscos movimientos fetales a modo de sacudidas y en otros de hipomotilidad intensa.

Las exploraciones llevadas a cabo a lo largo del embarazo para determinar la verdadera afectación implicaron el aporte de varios especialistas para su realización, interpretación y correcta evaluación: ecocardiograma, dilatación ligera de cavidades derechas; electromiograma, descargas miotónicas persistentes; hormonas tiroideas, dentro del rango de la normalidad; estudio oftalmológico, no aprecia cataratas ni favosclerosis; enzimas cardiacas, en el rango de la normalidad; exploración funcional respiratoria, resistencia de las vías aéreas ligeramente aumentadas y discreta pérdida de unidades alveolares para el intercambio y presiones respiratorias estáticas máximas disminuidas por pérdida de fuerza en la musculatura respiratoria. A lo largo del embarazo se objetiva aumento de la sintomatología que impide progresivamente la deambulación en especial en el tercer trimestre.
A la 37ª semana debido a la gran debilidad de los músculos abdominales, se decide en colaboración con los servicios de pediatría, neurología y anestesia finalizar la gestación mediante cesárea electiva con bloqueo intradural. Nace una niña de 2.500g, Apgar 3/7. Ph umbilical 7,30. Intervención satisfactoria y sin complicaciones, resaltando como hallazgo intraoperatorio de interés cordón umbilical corto. Puerperio con evolución favorable y progresiva mejoría del cuadro clínico.
El neonato con diagnóstico de DMC presentó como datos clínicos hipotonía intensa, píe equino-varo, ventriculomegalia cerebral, hipotonía pulmonar que precisó de respiración asistida prolongada y elevación de las cúpulas diafragmáticas.
DISCUSION
El presente caso de DMA ilustra su clásico inicio en la adolescencia, la típica sintomatología de miotonía y debilidad principalmente y su relación con los antecedentes familiares puesto que no se ha demostrado ninguna mutación nueva12. De igual manera, observamos en el curso del embarazo exacerbación de sus síntomas y posterior mejoría en el puerperio, datos que se corresponden con lo reportado por otros autores7.
La necesidad de formación de un equipo multidisciplinario se pone de manifiesto al considerarlo gestación de alto riesgo obstétrico, con posibilidad de descompensación cardiaca causada por una cardiomiopatía subclínica, fallos respiratorios por la exacerbación de la debilidad muscular tanto del diafragma como de los músculos intercostales13, requerimientos de evaluaciones neurológicas periódicas, mayor riego en los procedimientos anestésicos, así como necesidad de una asistencia perinatal calificada por la alta morbilidad y gravedad neonatal ante la posible DMC14.
Entre las complicaciones en el embarazo debemos mencionar el aumento en la tasa de abortos, desprendimiento prematuro de la placenta normoincerta, partos prematuros, disdinamias, ruptura prematura de membranas, presentaciones anómalas, así como el polihidrmanios, este último debido a la imposibilidad de deglutir líquido amniótico por parte del feto por la afección muscular imperante4.
Para el diagnóstico prenatal de DMC hemos recurrido a la amniocentesis pero desafortunadamente el rendimiento de los amniocitos fue escaso, pero sospechamos su presencia guiándonos en los datos clínicos de hipomotilidad fetal y ecográficos, polihidramnios, labios superiores en forma de signo circunflejo, pie equino-varo, hiperextensión de los miembros y desviación cubital del primer dedo de ambos pies, datos confirmados posteriormente al nacimiento y todos ellos reportados por diversos autores15-17. Además pueden objetivarse ecográficamente hidrops fetal no inmune, postura anormal de las manos y boca en tienda de campaña18,19.
Por otra parte, cualquiera de las tres fases del parto pueden estar afectadas, por alteración del miometrio que provocaría contracciones irregulares y lenta progresión en la dilatación cervical, corregido mediante la administración de oxitocina o bien por incontinencia funcional provocar un desenlace excesivamente rápido13. La asistencia del periodo expulsivo suele inducir un parto instrumental por la reducción de la capacidad tanto de la musculatura miometrial como abdominal y finalmente destacar la posibilidad de hemorragias puerperales o bien retención placentaria por acretismo20.
La elección tanto del momento como de la vía de finalización de la gestación debe ser considerada desde el punto de vista obstétrico y clínico, idealmente previo consenso entre el medico ginecólogo - obstetra, neurólogo y pediatra, así como la decisión del tipo de anestesia a ser utilizada debe ser discutida detalladamente entre todos los profesionales junto al anestesista, debido a la afectación multisitémica de esta patología, que inducen a considerar la posibilidad de mayores complicaciones durante el acto quirúrgico (sobre todos retención de secreciones por reducción de las presiones respiratorias) así como en el post-operatorio (atelectasias y bronconeumonías por aspiración).
En tal sentido, la cesárea es la vía elegida la mayoría de las veces debido a las anomalías en las presentaciones así como a la probable disfunción de la contractilidad13 y la combinación de técnicas espinales y epidurales la escogida como soporte anestésico en vista de que está contraindicado la anestesia general por las potenciales problemas que puede acarrear el uso de relajantes musculares y fármacos depresores del sistema nervioso central21.
Finalmente destacar que los antecedentes familiares, la clínica materna, los signos ecográficos y la no percepción materna de movimientos fetales hicieron sospechar la DMC fetal en virtud del diagnóstico prenatal no informativo. Así mismo, el manejo de esta gestación con un equipo formado por numerosos especialistas como neurólogos, pediatras, internistas, ecografistas, genetistas, anestesistas, entre otros, permitió valorar suficientemente la afección actual, su repercusión en el embarazo, ofrecer información adecuada a la pareja para determinar su prosecución, lo que benefició la correcta planificación de la asistencia perinatal.
BIBLIOGRAFIA
1. Urbano-Márquez A, Grau Junyent JM, Casademont Pou J, Cardellach López F. Enfermedades musculares En: Farreras / Rozman. Medicina Interna. 13º edición. Barcelona: Mosby Doyma Libros SA; 1996.p.1567-74. [ Links ]
2. Darras BT, Jones Jr HR. Diagnosis of pediatric neuromuscular disorders in the era of DNA analysis. Pediatr Neurol 2000; 23:289-300. [ Links ]
3. Jordan García J, Fernández López A, Romera Modamio G, Rodríguez Miguélez JM, Ballesta F, Figueras Aloy J. Distrofia congénita miotónica de Steinert. Aspectos genéticos. An Esp Pediatr 1997; 47: 539-42. [ Links ]
4. Barber MA, Eguiluz I, Plasencia W, Ramírez O. Distrofia miotónica de Steinert y gestación. Clin Invest Gin Obst 2003; 30:191-5. [ Links ]
5. Reardon W, Newcombe R, Fentol I, Sibert J, Harper PS. The natural history of congenital myotonic dystrophy: mortality and long term clinical aspects. Arch Dis Child 1993; 68:177-81. [ Links ]
6. Nonaka I, Kobayashi O, Osari S. Nondystrophinopathic muscular dystrophies including myotonic dystrophy. Sem Pediatr Neurol 1996; 3:100-21. [ Links ]
7. Dufour P, Berard J, Vinatier D, Savary JB, Dubreucq S, Monnier JC, et al. Myotonic dystrophy and pregnancy. A report of two cases and review of the literature. Eur J Obstet Gynecol Reprod Biol 1997; 72:159-64. [ Links ]
8. Fossen D, Gjertad L. Obstetric complications as the first sign of myotonic dystrophy. Acta Obstet Gynecol Scand 1986; 65:667-8. [ Links ]
9. Martín P, Sierra J, Losada A, Rufo M, Lucas A. Estudio genético molecular en la distrofia miotónica congénita. Rev Neurol 1997; 25:833-6. [ Links ]
10. Zühlke C, Atici J, Martorell, Gembruch U, Kohl M, Göpel W, Schwinger. Rapid detection of expansions by PCR and non-radioactive hybridization: application for prenatal diagnosis of myotonic dystrophy. Prenat Diagn 2000; 20:66-9. [ Links ]
11. González de Dios J, Martínez Frías ML, Egües Jimeno J, Gairi Tahull JM, Gómez Sabrido F, Moráles Fernández MC, et al. Estudio epidemiológico de la distrofia miotónica congénita de Steinert: características dismorfológicas. An Esp Pediatr 1999; 51:389-96. [ Links ]
12. Harley HG. Expansion o fan instable DMA region and phenotypic variation in myotonic dystrophy. Nature 1992; 355:545-6. [ Links ]
13. Atlas I, Smolin A. Combined maternal and congenital myotonic dystrophy manager by a multidisciplinary team. Eur J Obstet Gynecol Rerpod Biol 1999; 87:175-8. [ Links ]
14. Rittler M, Feld V, Montagno M. Distrofia miotónica congénita. Rev. Hosp. Mat. Inf. Ramón Sardá 1997; XVI:34-40. [ Links ]
15. Delest A, Elhage A, Cosson M. Steinert´s disease and pregnancy. A case report and recent literature. J Gynecol Obstet Biol Reprod (París) 1995; 24:177-80. [ Links ]
16. Ito T, Tanikawa M, Miura H, Teshima N, Kadowaki K, Nagata N, et al. The movements of fetuses with congenital myotonic dystrophy in utero. J Perinat Med 1996; 24:277-82. [ Links ]
17. Risseeuw JJ, Oudshoorn JH, van der Straaten PJ, Kuypers JC. Myotonic dystrophy in pregnancy: a report of two cases within one family. Eur J Gynecol Obstet Reprod Biol 1997; 73:145-8. [ Links ]
18. Esplin MS, Hallam MD, Farrington PF, Nelson L, Byrne J, Ward K. Myotonic dystrophy is a significant cause of idiopathic polihydramnios. Am J Obstet Gyencol 1998; 179:974-7. [ Links ]
19. Marshiach R, Rimon E, Achiron R. Tent-shaped mouth as a presenting symptom of congenital myotonic dystrophy. Ultrasound Obstet Gynecol 2002; 20:312-3. [ Links ]
20. Freeman RM. Placenta acreta and myotonic dystrophy: two case reports. Br J Obstet Gynecol 1991; 594:5. [ Links ]
21. Rusell SH, Hirsh NP. Anaesthesia and myotonia. Br J Amaesth 1994; 72:210-6. [ Links ]

{kind=link}
{kind=link}
{kind=link}