REPORTE DE CASO
45,X/46,XY. Variante de Síndrome de Turner. Reporte de un caso
45,X/46,XY. A variation of Turner Syndrome. A case report
Norma Monjagata
1
Elodia Torres
1
Stella Rodríguez
1
Silvia Fernández
1
Estefana Estigarribia
1
1Departamento de Genética. Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción. Paraguay
RESUMEN
El síndrome de Turner es una anomalía cromosómica descrita por primera vez por el Dr. Henry Turner en 1938 que se manifiesta principalmente por talla baja, cuello ancho, pterigyumcolli, cubitus valgo e infantilismo sexual. Tiene una prevalencia de 1 en 1800 a 5000 recién nacidos vivos femeninos y se caracteriza por la ausencia total o parcial del segundo cromosoma X. Con las técnicas citogenéticas una gran variedad de presentaciones han sido reconocidas, siendo la más común la monosomía del cromosoma X (constitución cromosómica: 45,X) y los menos frecuentes los mosaicismos, entre los que se incluyen cromosomas marcadores que corresponderían a fragmentos o la totalidad de un cromosoma Y; la presencia de este cromosoma podría conferirle al paciente características fenotípicas masculinas. Se reporta el caso de una niña de 14 años de edad con fenotipo de síndrome de Turner que presentó una constitución cromosómica en mosaico 45,X/46,XY. Madre y padre de 32 años, no consanguíneos, la niña fue traída a la consulta por ausencia de vello axilar y pubiano, y ausencia de desarrollo mamario En el nacimiento la paciente presentó genitales ambiguos, labios abiertos, en bolsa derecha el testículo se presentó atrofiado y el izquierdo en pelvis, ambos fueron extirpados a los 3 y 6 meses de vida respectivamente. Se realiza una revisión de la literatura y se propone el asesoramiento genético adecuado a lo hallado en el cariotipo.
Palabras clave: Síndrome de Turner; mosaicismo; monosomía; cariotipo; fenotipo
ABSTRACT
Turner syndrome (TS) is a chromosomal disorder discovered by Dr. Henry Turner in 1938, is manifested clinically mainly by short stature, broad neck, pterigyumcolli, cubitus valgus and sexual infantilism. It has a prevalence of 1 in 1800-5000 female live births and is characterized by the total or partial absence of the second X chromosome. A great variety of presentations have been recognized due to cytogenetic techniques, the most common being the monosomy of the X chromosome (chromosomal constitution: 45,X) and the less frequent mosaicism, including marker chromosomes that correspond to fragments or the whole Y chromosome. The presence of this chromosome could confer male phenotypic characteristics to the patients. We report the case of a 14-year-old girl with a phenotype similar to Turner syndrome who presented a mosaic chromosomal constitution 45 X/46,XY. Both parents were 32-year old, nonconsanguineous; the child was brought to consultation for absence of axillary and pubic hair and absence of breast development. At the birth the patient presented ambiguous genitalia, open labia and atrophied right testicle while the left remained in the pelvis, both were extirpated at 3 and 6 months of life respectively. A review of the literature was carried out and we proposed genetic counseling appropriate to the findings in the karyotype.
Keywords: Turner syndrome; mosaicism; monosomy; karyotype; phenotype
INTRODUCCIÓN
El síndrome de Turner(ST), es una patología cromosómica descrita por primera vez por el Dr. Henry Turner en 1938, que se manifiesta clínicamente por talla baja, cuello ancho, pterigyumcolli, cubitus valgo e infantilismo sexual1. Tiene una prevalencia de 1 en 1800 a 5000 recién nacidos vivos femeninos y se caracteriza por la ausencia total o parcial del segundo cromosoma X. Actualmente con las técnicas citogenéticas, una gran variedad de presentaciones han sido reconocidas, siendo la más común la monosomía del cromosoma X, cariotipo 45,X, y los menos frecuentes los mosaicismos, entre los que se incluyen cromosomas marcadores que corresponderían a fragmentos o la totalidad de un cromosoma Y2, este último tipo con una incidencia de 1,7 por cada 10.000 recién nacidos (XX2).
El mosaicismo se refiere a una condición en donde un individuo tiene dos o más poblaciones de células que difieren en su composición genética3. El desarrollo intersexual fue por primera vez observado cuando un porcentaje de células contenían cromosoma Y, como el caso de un mosaico45,X/46,XY1. La constitución cromosómica 45X/46,XY puede dar lugar a una amplia variedad de fenotipos, que abarca desde mujeres con estigmas de Síndrome de Turner, varones o mujeres con pseudohermafroditismo hasta varones fenotípicamente normales. Cuando un cromosoma Y está presente en mosaicismo en pacientes con ST, existe un riesgo incrementado de 15-25% de desarrollar gonadoblastoma y disgerminoma en la glándula disgenésica, motivo que conduce a la gonadectomía profilática. El manejo de pacientes con ambigüedad sexual incluye la asignación del sexo, gonadectomía, genitoplastia y lo relacionado con las características clínicas del Síndrome de Turner derivados de la línea celular 45,X4.
CASO CLINICO
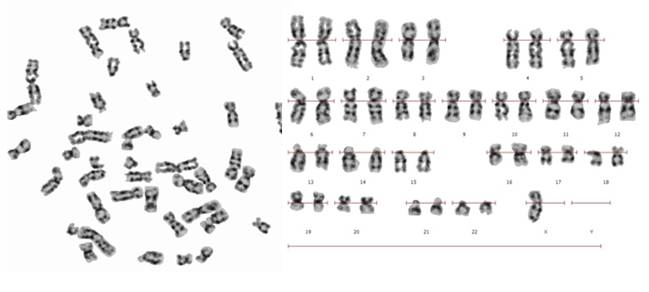
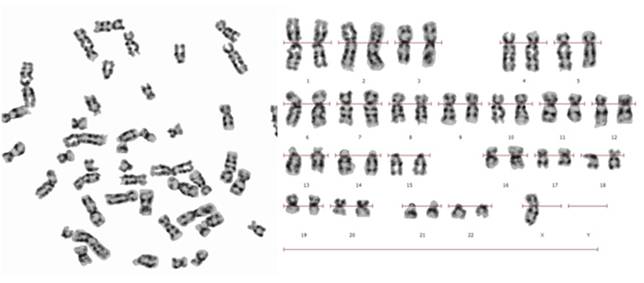
Se reporta el caso de una niña de 14 años de edad que concurrió a una consulta genética por ausencia del desarrollo de las glándulas mamarias, así como de vello axilar y pubiano, la paciente es hija de una pareja aparentemente sana no consanguínea. No se cuenta con los datos de procedencia. La madre refiere que la paciente nació con genitales ambiguos, aunque no aportó muchos datos al respecto, indico que por ecografía se observó en bolsa derecha un testículo, el cual fue extirpado a los 3 meses. Posteriormente se detectó el testículo izquierdo localizado en la pelvis, que fue extirpado a los seis meses. Al examen físico: CC: 52 cm (Percentil 50), Talla: 1,38 (Percentil< p 3) y cm Peso: 53 kg (Percentil: 50-75), no tiene desarrollo de mamas. El análisis cromosómico se realizó en linfocitos de sangre periférica. Fueron analizadas 50 metafases con bandas G y C, en las cuales se observó en un 54% una monosomía para el cromosoma X, y en el 46% restante se observó la presencia de cromosoma X e Y.
Cariotipo: 45,X/46,XY en mosaico (Figura 1 y Figura 2).
DISCUSIÓN
El síndrome de Tuner en sí es infradiagnosticado a pesar de su alta frecuencia de 1 por 1.800 a 5.000 recién nacidos del sexo femenino. La edad de consulta del caso presentado es un reflejo de lo que normalmente ocurre y explica el parte el motivo de la falta de diagnóstico, al cual se suma que casos como el descripto tiene una incidencia de 1,7 por cada 10.000 recién nacidos. Si bien no parece existir una asociación entre el grado del mosaicismo y la ocurrencia de defectos a nivel de los genitales externos, existe muy poca literatura que correlacione el fenotipo con el genotipo de este tipo de pacientes, pudiendo cursar con defectos como hipospadias, micropene, fusión de las bolsas u otros defectos, aunque se estima que el 60% cursan con genitales normales y un fenotipo masculino normal. Generalmente, los casos son descubiertos cuando se solicita el estudio cromosómico por la talla baja. Al revisar la literatura, se encontró que sólo un estudio se había realizado en América Latina para evaluar las características clínicas y citogenéticas de los pacientes con 45,X/46,XY mosaicismo9. Este fue desarrollado por Rosenberg et al7 quienes evaluaron los hallazgos fenotípicos de cinco pacientes. Otros estudios se desarrollaron en Europa2,8 pero especialmente en Estados Unidos10. Las referencias Nros. 5 y 6 deben aparecer antes de la 7 y 10.
La presentación de este caso tiene particular importancia para hacer un llamado a los profesionales de la salud sobre la necesidad del estudio cromosómico ante la presencia de un recién nacido con ambigüedad sexual tanto para la extirpación de las gónadas ante la posibilidad de malignización de estos como para la correcta asignación del sexo5. A pesar de que las anomalías de los cromosomas sexuales no cursan con discapacidad intelectual, estos sí tienen implicancia para el desarrollo de los caracteres sexuales secundarios y son en gran parte responsables de las disgenesias gonadales6.
Ante la presencia de una persona con 45,X/46,XY, se debe llevar a cabo una evaluación clínica exhaustiva similar a la realizada en las niñas con síndrome de Turner, para proceder a la extirpación de las gónadas si fuese necesario por el riesgo de gonadoblastoma que incluso puede presentarse en individuos con fenotipo y genitales normales pero con la mencionada constitución cromosómica11. La posibilidad de una respuesta favorable al tratamiento con hormonas de crecimiento y el apoyo sicológico, crucial tanto para el individuo afectado como para la familia, en el manejo de los casos de baja estatura o infertilidad son motivos adicionales para el diagnóstico precoz.
REFERENCIAS BIBLIOGRAFICAS
1. Tamar SC, Contreras NC, Fonseca DJ. Utilidad de la Citogenética en la medicina actual. Visión Histórica y Aplicación. Educación y Practica de la Medicina. [internet]. Acta Medica Colombiana, 2008; 33 (4). 309-16. [citado: 04 de agosto del 2017] Disponible en: Disponible en: http://www.actamedicacolombiana.com/cont.php?id=24&id2=162
[ Links ]
2. Saldarriaga Gil W, Ávila Sánchez F, Isaza C. Síndrome de Turner con mosaicismo 45X/46XY: Reporte de caso. Rev Chil Obstet Ginecol. [internet] 2011; 76(1):47-51. [citado: 07 de agosto del 2017] Disponible en: Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0717-75262011000100010
[ Links ]
3. Stankiewicz P, Lupski JR. Gene, Genomic, and Chromosomal Disorders. [ internet] In: Goldman L, Schafer AI, eds. Cecil Medicine. 24th ed. Philadelphia, Pa: Saunders Elsevier; 2011. [citado: 04 de agosto del 2017] .Disponible en: Disponible en: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/001317.htm
.
[ Links ]
4. Gordillo-González G, Escorcia P, Salvatierra I, Osorio G, Escobar- Ospina A. Disgenesia gonadal mixta por monosomía en mosaico: reporte de caso. [internet] Iatreia Revista Médica Universidad de Antioquia. 23(4). [citado 04 de agosto del 2017]. Disponible: Disponible: www.iatreia.udea.edu.co
[ Links ]
5. Richter-Unruh A, Knauer-Fischer S, Kaspers S, Albrecht B, Gillessen-Kaesbach G, Hauffa BP. Short stature in children with an apparently normal male phenotype can be caused by 45,X/46,XY mosaicism and is susceptible to growth hormone treatment.[ internet] Eur J Pediatr. 2004; 163(4-5): 251-6. [citado 04 de agosto del 2017]. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/14986122
[ Links ]
6. Machado-Rosa RF, Bartel-D'Ecclesiis WF, Papandreus-Dibbi R, Cardoso-Manique R, Trevisa P, Graziadio C, et al. Mosaicismo 45, X/46,XY: relato de 14 pacientes de um hospital do Brasil. Um estudo retrospectivo. [internet] Sao Paulo Med J. 2014; 132(6): [citado 07 de agosto del 2017]. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/23422775. Doi: 10.1590/1516-3180.2014.1326729
[ Links ]
7. Rosenberg C, Frota-Pessoa O, Vianna-Morgante AM, Chu TH. Phenotypic spectrum of 45,X/46,XY individuals. [internet] Am J Med Genet. 1987; 27(3):553-9. DOI: 10.1002/ajmg.1320270308. [citado 07 de agosto del 2017] Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/labs/articles/3631129/
[ Links ]
8. Telvi L, Lebbar A, Del Pino O, Barbet JP, Chaussain JL. 45,X/46,XY mosaicism: report of 27 cases. Pediatrics. [internet] 1999; 104(2 Pt 1): 304-8. [citado 07 de agosto del 2017]. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/10429013
[ Links ]
9. Tosson H, Rose SR, Gartner LA. Description of children with 45,X/46,XY karyotype.[ internet] Eur J Pediatr. 2012;171(3):521-9. [Ultimo acceso: 07 de agosto del 2017]. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/21997800
[ Links ]
10. Knudtzon J, Aarskog D. 45, X/46,XY mosaicism. A clinical review and report of ten cases. [ internet] Eur J Pediatr. 1987; 146(3):266-71. [Ultimo acceso: 07 de agosto del 2017]. Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/3595646
[ Links ]
11. Gantt PA, Byrd JR, Greenblatt RB, McDonough PG. A clinical and cytogenetic study of fifteen patients with 45, X/46,XY gonadal dysgenesis. [ internet] Fertil Steril. 1980; 34(3):216-21. [Ultimo acceso: 07 de agosto del 2017] Disponible en: Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/7409242
.
[ Links ]












 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink