










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
On-line version ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud vol.14 no.2 Asunción Aug. 2016
https://doi.org/10.18004/Mem.iics/1812-9528/2016.014(02)17-024
Articulo Original/ Original Article
Análisis filogenético y de presión evolutiva de secuencias nucleotídicas del gen VP4 de especies de enterovirus humanos
Phylogenetic and evolutive pressure analyses of nucleotide sequences of VP4 gene of human enterovirus species
Alina Acosta CabelloI, Graciela Russomando II, Emilio E. Espínola II**
IFacultad de Ciencias Químicas, Universidad Nacional de Asunción. Paraguay
IIDepartamento de Biología Molecular y Biotecnología, Instituto de Investigación en Ciencias de la Salud, Universidad Nacional de Asunción. Paraguay
* AAC presentó parte de este estudio como trabajo de grado, para obtener el título de Bioquímica
R E S U M E N
El género Enterovirus es un grupo viral que afecta a un amplio rango de hospederos, entre ellos los humanos (especies A, B, C, y D), causan enfermedades respiratorias, gastrointestinales, neurológicas, y otras, y son altamente contagiosos. Los síntomas pueden ser leves o graves. El objetivo del trabajo fue analizar la variación nucleotídica, filogenética y de presión evolutiva de secuencias nucleotídicas del gen VP4 de las cuatro especies que afectan a los humanos. Se emplearon 92 secuencias nucleotídicas disponibles en la base de datos GenBank; éstas se editaron con el software BioEdit y se alinearon con Clustal W; las relaciones filogenéticas se determinaron con MEGA6, y las presiones evolutivas con los algoritmos SNAP y SLAC. Se encontró que la identidad nucleotídica mínima intra-especie fue de 43,2% (especie B) a 72,6% (especie D). Los genotipos más variables por especie fueron EV-71 (A), Echovirus 2 (B), EV-118 (C), y EV-94 (D). El análisis de presión evolutiva mostró que el gen VP4 en las cuatro especies evoluciona bajo presión selectiva negativa. Esto indicaría que la alta tasa mutacional y eventos de recombinación no tienen un rol significativo en la evolución de este gen, debido probablemente a la localización interna de la proteína VP4.
Palabras claves: Enterovirus humanos, secuencias nucleotídicas, gen VP4, análisis filogenético, presión selectiva.
A B S T R A C T
The Enterovirus genus is a viral group that affects a wide host range, including humans (species A, B, C and D), cause respiratory, gastrointestinal, and neurologic disease, among others, and are highly contagious. The symptoms range from mild to severe. The objective of this study was to perform a nucleotidic variation, phylogenetic and selective pressure analyses of the VP4 gene from the four enterovirus species that affect humans. Ninety-two nucleotide sequences (available in the GenBank database) were employed; they were edited with BioEdit software and aligned with Clustal W; the phylogenetic relationships were determined with MEGA6, and the evolutive pressures with SNAP and SLAC algorithms. It was found an intra-species nucleotide identity of at least 43,2% (species B) to 72,6% (species D). The more variable genotypes by species were EV-71 (A), Echovirus 2 (B), EV-118 (C), and EV-94 (D). The selective pressure analysis showed that VP4 gene of the four species evolves by negative pressure. This would indicate that the high mutation rate and recombination events do not have a significant role in the evolution of this gene, probably due to the internal localization of the VP4 protein.
Key words: Human enteroviruses, nucleotide sequences, VP4 gene, phylogenetic analysis, selective pressure.
INTRODUCCCIÓN
Los Enterovirus, pertenecientes a la familia Picornaviridae, comprenden un grupo de virus de tamaño pequeño y RNA como material genético (1). Los viriones son esferoidales, con un diámetro de aproximadamente 30 nanómetros. La cápside es simétrica y posee un arreglo de 60 subunidades llamadas protómeros, dispuestas en una estructura icosaédrica altamente empaquetada; cada protómero está formado por 4 proteínas estructurales denominadas proteínas virales (VP). En la superficie, cada bloque está constituido por VP1, VP2 y VP3, y en la parte interna están relacionadas con VP4 (1). El RNA viral es infeccioso porque una vez dentro de la célula huésped genera todas las proteínas virales requeridas para la replicación viral (1).
Se clasifican en 10 especies: Human enterovirus A, B, C y D; Simian enterovirus, Bovine enterovirus, Porcine enterovirus B; y Human rinhovirus A, B y C. La clasificación se realiza en función a la identidad aminoacídica de proteínas externas compartida por los miembros de una especie; por lo tanto, cada especie enteroviral comprende características en común compartidas por los miembros que los conforman (1).
Los Enterovirus humanos no se limitan a afectar exclusivamente el tracto gastrointestinal o respiratorio; también pueden producir infecciones del sistema nervioso central (meninges y encéfalo), páncreas, corazón y también se los han relacionado con la aparición de diabetes mellitus (2).
La variabilidad genética es una determinante importante en la generación de nuevos tipos o variantes virales. Entre los principales mecanismos generadores de variabilidad en virus RNA se encuentran las mutaciones y recombinaciones. Las mutaciones nucleotídicas pueden darse debido a la incapacidad de la RNA polimerasa de corregir errores durante la replicación, lo que ocasiona elevadas tasas de mutaciones por sustitución (3). La recombinación, originada por intercambio genético entre dos cepas virales al infectar una misma célula, puede originar cepas con características muy distintas a las predecesoras; este mecanismo, aparte de contribuir a la variabilidad genética y la generación de nuevos virus, reduce la carga mutacional (4, 5).
La presión selectiva se define como el grado con que los organismos son favorecidos o desfavorecidos por las mutaciones genéticas (6), pudiendo ocasionar dos escenarios posibles: 1) el cambio en el codón no produce cambio en el aminoácido codificado (denominado sustitución sinónima), y 2) el cambio en el codón produce cambio del aminoácido (denominado sustitución no sinónima). Una presión selectiva positiva implica que la variación aminoacídica que sufrió el gen, favoreció a la especie en su supervivencia. La presión negativa, en cambio, se refiere a que las condiciones de selección negativa para ese gen han permanecido iguales, eliminando los cambios que favoreciesen a la especie (7).
Las reconstrucciones filogenéticas son utilizadas como una herramienta útil para el análisis evolutivo de secuencias génicas, permitiendo la clasificación de los genes estudiados de manera más sencilla y relacionándolos con factores evolutivos de la especie y la historia demográfica de las poblaciones en estudio. Además, se puede obtener información sobre la dinámica de los virus a partir de la estructura de las ramas en un árbol filogenético (8).
En América Latina, existen datos de circulación de las cuatro especies de enterovirus humanos, en mayor porcentaje los genotipos de la especie B (9). En Paraguay no existen datos precisos sobre especies ni genotipos enterovirales asociados a manifestaciones clínicas, por lo que este estudio podría utilizarse como punto de partida para análisis moleculares de enterovirus de importancia en Salud Pública en nuestro país.
MATERIALES Y MÉTODOS
Obtención de secuencias nucleotídicas
Se obtuvieron en total 92 secuencias nucleotídicas de referencia del gen de VP4 de enterovirus humanos (especies A, B, C, y D), de la base de datos GenBank (http://www.ncbi.nlm.nih.gov); cada gen estaba conformado por 67 codones (201 nucleótidos); cada secuencia correspondió a un genotipo representativo por especie, según asignación taxonómica en la base de datos de los picornavirus (http://www.picornaviridae.com/). El número de secuencias obtenidas por especie fue de 17 para la especie A, 55 para la B, 17 para la C y 3 para la D.
Análisis filogenético y de identidad nucleotídica
Las secuencias nucleotídicas obtenidas fueron manualmente editadas con BioEdit v.7.0.5 (10). El alineamiento múltiple de secuencias fue realizado con Clustal W (11). La reconstrucción filogenética fue realizada con el método de neighbor-joining, utilizando el método de Kimura 2-parámetros como modelo de sustitución nucleotídica, y análisis de bootstrap de 1.000 réplicas con el programa MEGA v6 (12). Para el cálculo de identidad nucleotídica, se realizó un análisis de distancia entre pares de secuencias (pairwise distance) dentro de una misma especie, utilizando el programa MEGA v6 (12); a partir de las distancias, se dedujeron las identidades (expresadas en porcentajes).
Análisis de las presiones selectivas
Para el análisis de presión selectiva, se utilizó el método de Nei y Gojobori (7). Los datos fueron analizados utilizando el programa SNAP (13), basado en el lenguaje de programación Perl; para ello, se partió de un alineamiento múltiple de secuencias, y se contabilizó el número de cambios sinónimos y no sinónimos para cada codón. Al hallar la relación entre las sustituciones no sinónimas (dn) y sinónimas (ds), i.e. dn/ds, si el valor es 1, la presión de selección es neutra; si el valor es menor a 1 implica una presión de selección negativa, y si es mayor a 1, una presión selectiva positiva (7).
La determinación del número de codones bajo presión selectiva positiva, negativa o neutra se estimó con el algoritmo “Single likelihood ancestor counting” (SLAC) de la interfase Datamonkey (14).
RESULTADOS
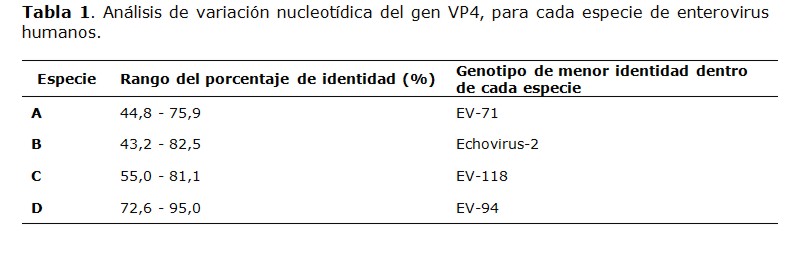
Se encontró que la identidad nucleotídica mínima intra-especie fue de 43,2% (especie B) a 72,6% (especie D) (Tabla 1). Los genotipos más variables por especie fueron EV-71 (A), Echovirus 2 (B), EV-118 (C), y EV-94 (D) (Tabla 1).
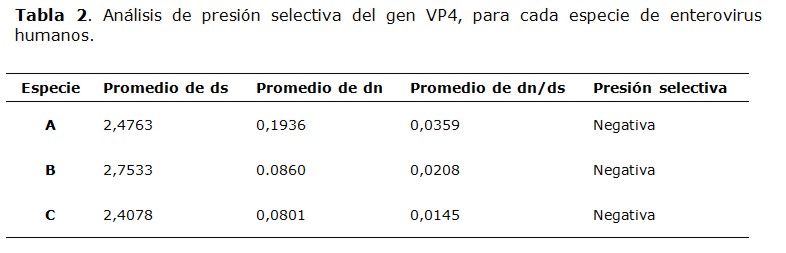
El análisis SNAP de presión selectiva de enterovirus humanos de las especies A, B, y C demostró que el gen VP4 evoluciona mediante presión selectiva negativa, con tasas dn/ds que van de 0,0145 hasta 0,0359 (Tabla 2). No se realizó el análisis de la especie D debido al escaso número de secuencias disponibles (se necesitan como mínimo tres secuencias de genotipo distinto). Mediante el algoritmo SLAC, se determinó que el número de codones bajo presión selectiva negativa fue de 66/67 codones; no se observaron codones bajo presión positiva.
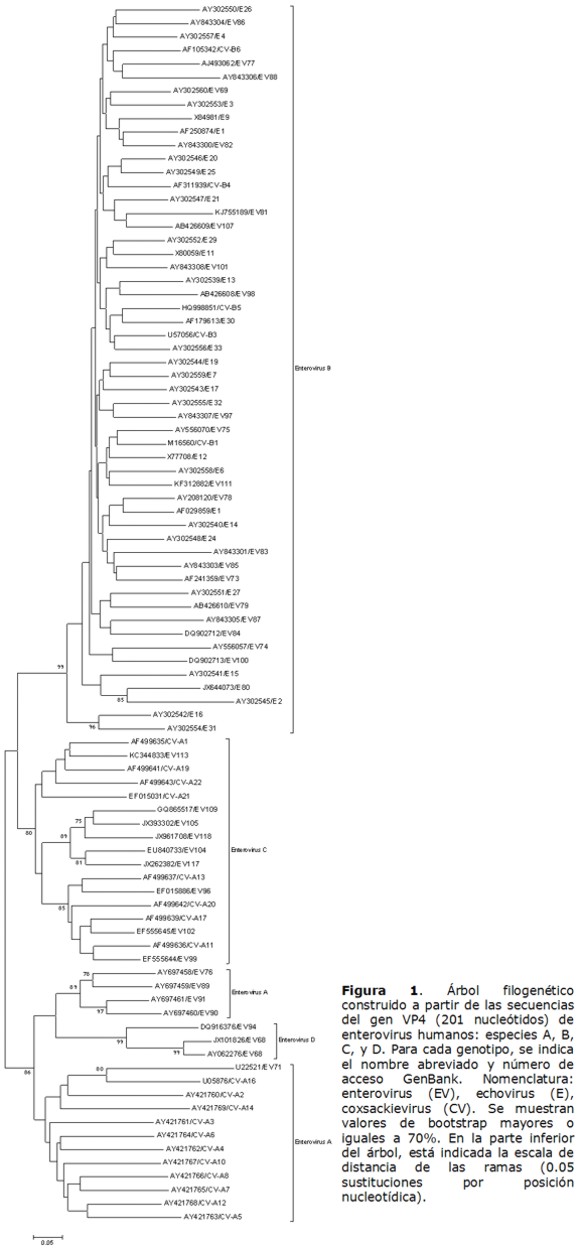
La relación entre las cuatro especies de enterovirus humanos por análisis filogenético (Figura 1) demostró la relación entre Enterovirus A y Enterovirus D, debido al agrupamiento de estas especies en un único cluster, con un valor bootstrap de 86%. Los genotipos de Enterovirus B y Enterovirus C formaron clusters separados (Figura 1).
DISCUSIÓN
En los últimos años han aumentado las aplicaciones de técnicas bioinformáticas para comprender la epidemiología molecular de las enfermedades ocasionadas por enterovirus; por ejemplo, mediante tales herramientas se descifró el genoma de diversos enterovirus (15), se identificaron las regiones genómicas de importancia funcional, se determinaron las relaciones genéticas entre las diferentes cepas (16),así como también se reportaron mecanismos probables de generación de variabilidad genética y evolución viral (17, 18).
Este trabajo se ha enfocado en el análisis del gen codificante de la proteína VP4 de genotipos representativos de enterovirus humanos, debido a que esta proteína tiene importancia estructural para la cápside viral (19), está estrechamente relacionada al genoma viral (20), y por poseer una secuencia conservada entre los enterovirus.
En el análisis de variación nucleotídica intra-especie, se observaron porcentajes mínimos de identidad de 43,2% (especie B) a 72,6% (especie D). El bajo porcentaje de identidad nucleotídica dentro de la especie B se debería a eventos de recombinación genética intra-especie entre genotipos relacionados (5); dentro de esta especie, el genotipo de menor identidad nucleotídica correspondió al echovirus 2, que son altamente infecciosos, sobre todo en pacientes pediátricos, lo que favorece la aparición de brotes y por consiguiente el incremento de la tasa mutacional (21). El alto porcentaje de identidad nucleotídica dentro de la especie D podría deberse a que las cepas pertenecientes a este grupo son de circulación reciente, probablemente derivados de la especie A (según nuestro análisis filogenético); dentro de la especie D, EV-94 fue el genotipo de menor identidad nucleotídica, el cual muestra altas tasas mutacionales según estudios epidemiológicos a nivel mundial, y es relativamente nuevo con respecto a los demás genotipos de la especie D (22, 23) . En cuanto a la especie A, el genotipo de menor identidad fue el EV-71, que está asociado a diversas patologías, y no solo a problemas de boca, pies y manos; además, es el virus no-polio más dañino a nivel neuromotor (24). Fue comprobado que el EV-71 sufre cambios adaptativos en el genoma (25), sobre todo en la proteína VP1 que confiere resistencia a algunos inhibidores virales (26). Por último, dentro de la especie C, el genotipo de menor identidad fue el EV-118, correspondiente a un nuevo tipo enteroviral identificado dentro de esta especie; la región nucleotídica no traducida de EV-C118 es filogenéticamente distinta a la de los enterovirus humanos de la especie C clásicos; no obstante, está estrechamente relacionado con el EV-109 y el EV-105 (27-29). Esto también se aprecia en el árbol filogenético que obtuvimos en nuestro análisis, lo que podría sugerir un origen de recombinación y posible evolución independiente de las diferentes regiones genómicas del EV-118 (29).
En cuanto al análisis de los patrones de sustituciones sinónimas y no sinónimas, se observó que el gen VP4 de las especies de enterovirus humano A, B, y C, evoluciona por presión negativa. Esto indicaría que la alta tasa mutacional y eventos de recombinación no tienen un rol muy significativo en la evolución de este gen, debido probablemente a la localización interna de la proteína VP4, que no está bajo la presión selectiva del sistema inmune del hospedero. Esto sí ocurre con los genes codificantes de las proteínas expuestas en la superficie viral, tales como VP1 y VP2 (30-32).
En los últimos años se ha detectado la circulación de enterovirus humanos causantes de altas tasas de morbilidad y mortalidad en el mundo; por ejemplo, en 2014, se reportó en Estados Unidos de América un brote de enterovirus 68 (especie D) que causó enfermedad respiratoria severa en niños, por lo que es considerado como un patógeno reemergente (33, 34). Por esto, es importante establecer líneas de investigación que permitan identificar y genotipificar los enterovirus circulantes en el Paraguay.
REFERENCIAS BIBLIOGRAFICAS
1. Racaniello VR. Picornaviridae: the viruses and their replication. In: Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, et al., editors. Fields Virology. 1. Philadelphia: Lippincott Williams & Wilkins; 2013. p. 453-89. [ Links ]
2. Brown BA, Oberste MS, Alexander JP, Jr., Kennett ML, Pallansch MA. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. Journal of virology. 1999;73(12):9969-75. [ Links ]
3. Domingo E, Ruiz-Jarabo CM, Sierra S, Arias A, Pariente N, Baranowski E, et al. Emergence and selection of RNA virus variants: memory and extinction. Virus research. 2002;82(1-2):39-44. [ Links ]
4. Burke DS. Recombination in HIV: an important viral evolutionary strategy. Emerging infectious diseases. 1997;3(3):253-9. [ Links ]
5. Oberste MS, Maher K, Pallansch MA. Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. Journal of virology. 2004;78(2):855-67. [ Links ]
6. Eigen M, Gardiner W, Schuster P, Winkler-Oswatitsch R. The origin of genetic information. Scientific American. 1981;244(4):88-92, 6, et passim. [ Links ]
7. Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular biology and evolution. 1986;3(5):418-26. [ Links ]
8. Tanaka T, Nei M. Positive darwinian selection observed at the variable-region genes of immunoglobulins. Molecular biology and evolution. 1989;6(5):447-59. [ Links ]
9. Garcia J, Espejo V, Nelson M, Sovero M, Villaran MV, Gomez J, et al. Human rhinoviruses and enteroviruses in influenza-like illness in Latin America. Virology journal. 2013;10:305. [ Links ]
10. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95-8. [ Links ]
11. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673-80. [ Links ]
12. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular biology and evolution. 2013;30(12):2725-9. [ Links ]
13. Korber B. HIV Signature and Sequence Variation Analysis. Computational Analysis of HIV Molecular Sequences. Dordrecht, Netherlands: Kluwer Academic Publishers; 2000. [ Links ]
14. Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21(10):2531-3. [ Links ]
15. Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420-8. [ Links ]
16. Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, et al. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40(Database issue):D593-8. [ Links ]
17. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research. 2012;22(3):568-76. [ Links ]
18. Prachayangprecha S, Schapendonk CM, Koopmans MP, Osterhaus AD, Schurch AC, Pas SD, et al. Exploring the potential of next-generation sequencing in detection of respiratory viruses. Journal of clinical microbiology. 2014;52(10):3722-30. [ Links ]
19. Chow M, Newman JF, Filman D, Hogle JM, Rowlands DJ, Brown F. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature. 1987;327(6122):482-6. [ Links ]
20. Danthi P, Tosteson M, Li QH, Chow M. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. Journal of virology. 2003;77(9):5266-74. [ Links ]
21. Williams RC. The Probable Mutation Effect: neutral alleles and structural reduction. Human biology. 1978;50(2):173-81. [ Links ]
22. Smura TP, Junttila N, Blomqvist S, Norder H, Kaijalainen S, Paananen A, et al. Enterovirus 94, a proposed new serotype in human enterovirus species D. The Journal of general virology. 2007;88(Pt 3):849-58. [ Links ]
23. Smura T, Ylipaasto P, Klemola P, Kaijalainen S, Kyllonen L, Sordi V, et al. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. Journal of medical virology. 2010;82(11):1940-9. [ Links ]
24. Melnick JL. Enterovirus type 71 infections: a varied clinical pattern sometimes mimicking paralytic poliomyelitis. Reviews of infectious diseases. 1984;6 Suppl 2:S387-90. [ Links ]
25. Miyamura K, Nishimura Y, Abo M, Wakita T, Shimizu H. Adaptive mutations in the genomes of enterovirus 71 strains following infection of mouse cells expressing human P-selectin glycoprotein ligand-1. The Journal of general virology. 2011;92(Pt 2):287-91. [ Links ]
26. Chen TC, Liu SC, Huang PN, Chang HY, Chern JH, Shih SR. Antiviral activity of pyridyl imidazolidinones against enterovirus 71 variants. Journal of biomedical science. 2008;15(3):291-300. [ Links ]
27. Daleno C, Greenberg D, Piralla A, Scala A, Baldanti F, Principi N, et al. A novel human enterovirus C (EV-C118) identified in two children hospitalised because of acute otitis media and community-acquired pneumonia in Israel. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2013;56(2):159-62. [ Links ]
28. Piralla A, Daleno C, Scala A, Greenberg D, Usonis V, Principi N, et al. Genome characterisation of enteroviruses 117 and 118: a new group within human enterovirus species C. PloS one. 2013;8(4):e60641. [ Links ]
29. Tapparel C, Junier T, Gerlach D, Van-Belle S, Turin L, Cordey S, et al. New respiratory enterovirus and recombinant rhinoviruses among circulating picornaviruses. Emerging infectious diseases. 2009;15(5):719-26. [ Links ]
30. Kim SH, Lee HY, Jang YS. Targeted Delivery of VP1 Antigen of Foot-and-mouth Disease Virus to M Cells Enhances the Antigen-specific Systemic and Mucosal Immune Response. Immune network. 2013;13(4):157-62. [ Links ]
31. Lindberg AM, Andersson P, Savolainen C, Mulders MN, Hovi T. Evolution of the genome of Human enterovirus B: incongruence between phylogenies of the VP1 and 3CD regions indicates frequent recombination within the species. The Journal of general virology. 2003;84(Pt 5):1223-35. [ Links ]
32. Zaini Z, McMinn P. A single mutation in capsid protein VP1 (Q145E) of a genogroup C4 strain of human enterovirus 71 generates a mouse-virulent phenotype. The Journal of general virology. 2012;93(Pt 9):1935-40. [ Links ]
33. Imamura T, Oshitani H. Global reemergence of enterovirus D68 as an important pathogen for acute respiratory infections. Reviews in medical virology. 2015;25(2):102-14. [ Links ]
34. Nelson R. Outbreaks of enterovirus D68 continue across the USA. The Lancet Respiratory medicine. 2014;2(10):791. [ Links ]
Fecha de recepción:junio 2016. Fecha de aceptación: agosto 2016
Autor correspondiente: *Emilio Espínola. Instituto de Investigación en Ciencias de la Salud, Universidad Nacional de Asunción, Paraguay.
E-mail: emilioespinola@hotmail.com

