










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
versão On-line ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud v.9 n.2 Asunción dez. 2011
REPORTE DE CASOS
Leucemia mieloide aguda cont(8; 21)(q22; q22).Reporte de casos
Acute myeloid leukaemia witht (8; 21)(q22; q22). Case report
Espínola Cano AFI, Rodríguez MSI, Campos SI, Ferreira Nizza JAII, Noguera JIII, *Figueredo Thiel SJI
IDepartamento de Hematopatología y genética, Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción, Paraguay
IIHematología, Hospital de Clínicas, Facultad de Ciencias Médicas
Universidad Nacional de Asunción. Paraguay
IIIOncohematologia, Hospital General Pediátrico Niños de Acosta Ñu, Ministerio de Salud Pública y Bienestar Social. Reducto, San Lorenzo, Paraguay
RESUMEN
La leucemia mieloide aguda es una neoplasia hematopoyética caracterizada por la proliferación clonal de blastos inmaduros en médulas ósea interfiriendo con sus funciones normales. Tiene una supervivencia aproximada de 35% afectando principalmente a adultos mayores de 60 años y niños menores de un año y preferentemente al sexo masculino. Un hallazgo frecuente es la presencia de la translocación cromosómica t (8; 21) (q22; q22) que involucra a los genes RUNX1 y RUNX1T1. La detección de esta alteración tiene implicancia diagnóstica y pronóstica de la enfermedad. El objetivo de este trabajo es describir y reportar dos casos de leucemia mieloide aguda en pacientes masculinos de 14 y 24 años que presentaron clínica, laboratorio y morfología típicos de la enfermedad pero con edad de aparición no habitual, enfatizando además, el pronóstico bueno desde el punto de vista citogenético de esta translocación en ambos casos.
Palabras clave: leucemia mieloide, translocación genética, análisis citogenético, pronóstico.
ABSTRACT
Acute myeloid leukemia is a haematopoietic neoplasia characterized by clonal proliferation of immature blasts in bone marrow, interfering with its normal functions. Overall survival is about 35% affecting mainly male adults over 60 years old and infants under one year old. Genetic translocation t(8;21)(q22;q22) is a recurrent finding and involves RUNX1 and RUNX1T1 genes. The detection of this genetic translocation is relevant to the diagnosis and prognosis of the disease. The objective of this work is to report two cases of acute myeloid leukaemia in 14 and 24 years old male patients with typical clinical, laboratorial and morphological findings but with unusual appearing ages, emphasizing the good prognosis from the genetic point of view of this translocation in both cases.
Keywords: myeloid leukemia, genetic translocation, cytogenetic analysis, prognosis.
INTRODUCCIÓN
La leucemia mieloide aguda (LMA) es una neoplasia clonal maligna del tejido hematopoyético. Se caracteriza por la proliferación descontrolada de células blásticas inmaduras en médula ósea, sangre periférica y otros órganos, a los que infiltra interfiriendo con sus funciones (1). Esto se traduce en una disminución de la producción de células sanguíneas normales con los consecuentes síntomas asociados, como anemia severa, infecciones frecuentes y hemorragias, llegando a ser fatal dentro del primer año si no se trata adecuadamente (2). La enfermedad afecta frecuentemente a adultos mayores de 60 años y niños menores de un año, con una supervivencia aproximada del 35% (1,2). Clínicamente, pacientes con LMA presentan cansancio, debilidad, palidez, disnea de esfuerzo, petequias, epistaxis y hemorragia gingival y/o conjuntival. Un 25% presenta dolores óseos y articulares como síntomas iniciales, aproximadamente la mitad presenta esplenomegalia, siendo las adenomegalias infrecuentes al igual que la afectación del sistema nervioso central. El laboratorio muestra anemia moderada a severa, trombocitopenia marcada, leucocitosis a expensas de células blásticas con disminución absoluta y/o relativa de los demás componente normales (3). Pueden encontrarse además, recuentos normales, disminuidos o inclusive ausencia de blastos (formas aleucémicas) (4).
Morfológicamente la médula ósea, se caracteriza por presencia de grandes células blásticas, de linaje granulocítico que presentan freno madurativo o hiatus leucémico. Estos blastos poseen abundante citoplasma claro a basofílico, presencia de bastones de Auer y en algunas ocasiones con gránulos muy grandes de tipo Chediak-Higashi (5,6). El inmunofenotipo es inmaduro con expresión fuerte de CD34, HLA-DR y antiMPO, y expresión débil de CD33 (7). Estudios citogenéticos y moleculares demuestran un gran número de alteraciones genéticas en las LMA, siendo la más frecuente, la translocación cromosómica balanceada t(8;21)(q22;q22) (5,8). Este conjunto de alteraciones es producto de un desbalance genético producido por la fusión del gen RUNX1 (cromosoma 21), que regula directamente múltiples genes involucrados en el ciclo celular y en la diferenciación linfoide/mieloide de las células hematopoyéticas (9), con el gen RUNX1T1 (cromosoma 8) que produce la proteína ETO que posee segmentos de anclaje a varios co-represores y a histona desacetilasas (HDAC) (10). Cuando dichos genes están fusionados se produce el factor de trascripción conocido como AML1-ETO, una proteína que mantiene la capacidad de unirse al ADN e interactuar con inhibidores de la trascripción, lo que desencadena la falta de maduración y diferenciación en las células de este tipo de LMA (10-12).
A pesar de la gravedad que reviste un cuadro de LMA, la presencia de esta translocación en pacientes afectados, detectada ya sea por métodos citogenéticos o moleculares, está asociada a una buena respuesta al esquema de tratamiento actual, así como a una mayor sobrevida libre de enfermedad (2, 13, 14). Teniendo en cuenta que las alteraciones cromosómicas son detectables hasta en un 55% (8) de LMA, su identificación resulta de suma importancia para el diagnóstico y más aún, para el pronóstico, siendo imprescindibles los estudios citogenéticos y moleculares para mejorar el esquema terapéutico quimioterápico y el éxito del tratamiento de estos casos de buen pronóstico (2, 8, 15).
El objetivo de este trabajo es describir y reportar dos casos de leucemia mieloide aguda con translocación t (8;21) destacando que este aspecto, en conjunto con los hallazgos clínicos, morfológico-citoquímicos e inmunofenotípicos permitieron sub-clasificar a ambos pacientes como LMA de buen pronóstico.
PRESENTACIÓN DE CASOS
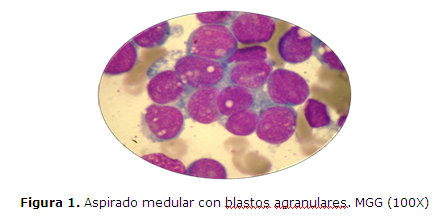
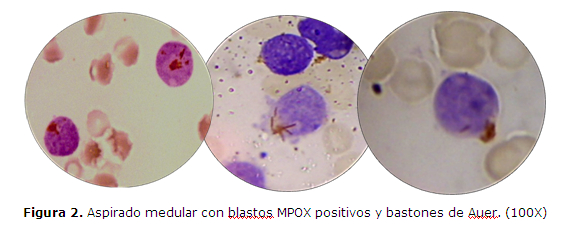
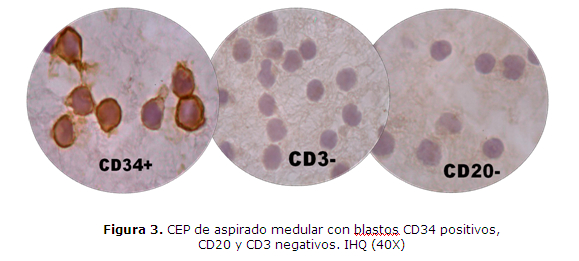
CASO CLINICO 1: Varón de 14 años, procedente de Choré, San Pedro, que consulta al Hospital General Pediátrico Niños de Acosta Ñu por esplenomegalia. Historia pre-hospitalaria de palidez de piel y mucosas, de moderada intensidad, de un mes de evolución, fiebre de 38ºC sin escalofríos ni sudoración, dolor en el pecho y vómito de contenido alimentario con estrías de sangre, 24hs antes del ingreso. Al examen físico presenta petequias dispersas en ambas piernas y pápulas en miembro inferior derecho. El laboratorio informa Leucocitos: 25.200/uL con 68% de blastos, Hemoglobina: 6,1g/dL, Hematocrito: 17%, Plaquetas: 31.000/uL. La anatomía patológica informa que en el aspirado de médula ósea se observa 92% de blastos granulares de tipo II, mieloperoxidasa (MPOX) positivos, de citoplasma escaso a moderado, con presencia de ocasionales bastones de Auer, núcleos de cromatina fina inmadura con uno a tres nucléolos evidentes (figura 1 y 2). El inmunofenotipo realizado con técnicas de inmunohistoquímica (IHQ) en cortes del coágulo medular procesado en parafina (CEP), evidencia expresión intensa de CD34 y antiMPOX, sin expresión de CD20 ni CD3 (figura 3). El estudio citogenético revela cariotipo 46,XY, t(8;21)(q22;q22) (figura 4). Se lo diagnóstica como LEUCEMIA MIELOIDE AGUDA SIN MADURACIÓN, LMA-M1 de FAB con t(8;21)(q22;q22) de OMS. El paciente es retirado del servicio al quinto día de internación sin recibir tratamiento a pedido expreso de los padres para ser tratado con medicina alternativa en su comunidad, a pesar de los esfuerzos médicos, falleciendo días después en otro centro asistencial.

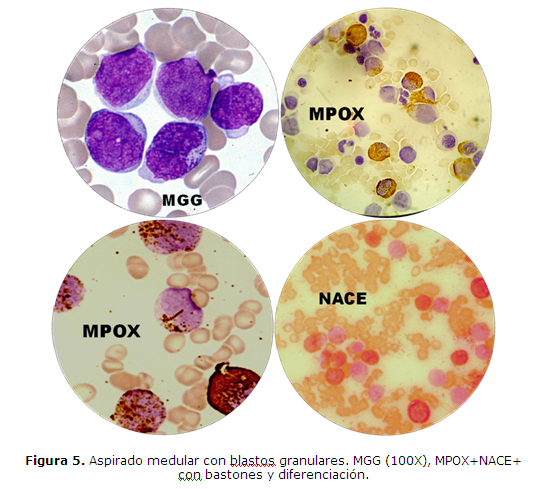
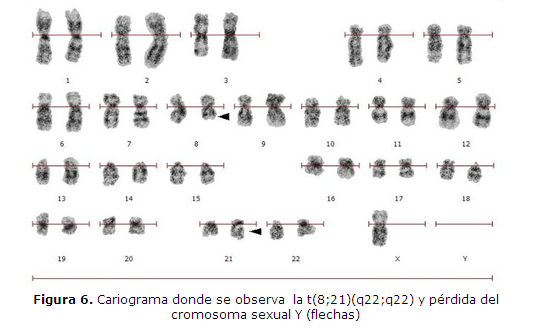
CASO CLINICO 2: Varón de 24 años, de profesión estibador, procedente de Campo 9, Caaguazú, con historia pre-hospitalaria de mareos, cefalea y disnea de esfuerzo progresiva, de tres semanas de evolución. Ingresa al Servicio de Urgencias del Hospital de Clínicas, Facultad de Ciencias Médicas-UNA, remitido del Hospital Regional de Ciudad del Este, presentando palidez de piel y mucosas, aparición espontánea de equimosis en miembros y tronco, sangrado espontáneo de encías y hepato-esplenomegalia. El laboratorio muestra Leucocitos: 11.800/uL con 65% de blastos, Hemoglobina: 10,0g/dL, Hematocrito: 30%, Plaquetas: 20.000/uL. La anatomía patológica informa médula ósea con 57% de blastos granulares de tipo II, mieloperoxidasa (MPOX) y alfa-naftol AS-D cloroacetatoesterasa (NACE) positivos, de citoplasma escaso a moderado, con bastones de Auer (figura 5) e inmunofenotipo con expresión acentuada de CD34 y antiMPOX, y expresión negativa de CD20 y CD3. El estudio citogenético presenta cariotipo 45, X,-Y, t(8;21)(q22;q22) (figura 6). Se lo diagnostica como LEUCEMIA MIELOIDE AGUDA CON MADURACIÓN, LMA-M2 de FAB con t (8;21)(q22;q22) de OMS. El paciente recibe el esquema quimioterápico correspondiente entrando en remisión completa luego de la fase de inducción. Durante la fase de consolidación ingresa al Servicio de Urgencias del Hospital por cuadro de shock séptico durante el cual fallece.
DISCUSIÓN
Siendo la leucemia mieloide aguda una neoplasia maligna hematológica que afecta principalmente a niños menores de un año y adultos mayores, con una incidencia en Estados Unidos y Europa de 1,6 casos/100.000 habitantes en menores de 60 años, elevándose a 17,9 casos/100.000 habitantes en personas mayores (16). En el Paraguay, un estudio realizado durante el periodo 1997-2002 en 370 pacientes hematológicos mostró una frecuencia de 12,7% de LMA, afectando mayormente a adultos (62%) y al sexo masculino (niños 61% y adultos 65%) (17).
Siguiendo los datos clásicos de la literatura, la incidencia de la LMA aumentaría con la edad, lo que no se ha encontrado en los casos reportados en este estudio que presentaron edades de 14 y 24 años. Sí, cabe destacar la coincidencia de frecuencia del género masculino y las manifestaciones clínicas iniciales de estos pacientes, que fueron las típicamente encontradas en este tipo de patología. Se observó palidez de piel y mucosas, disnea de esfuerzo, mareos y cefaleas, consecuencias de una anemia marcada, así como petequias, moretones y sangrados productos de la trombocitopenia severa. Estas citopenias, desencadenadas por la rápida proliferación de las células blásticas que invaden la médula ósea, le impiden llevar a cabo su función normal (1-4, 16). Los altos porcentajes de blastos encontrados en el hemograma y mielograma evidencian lo expuesto.
Para la clasificación de la enfermedad, estas fueron variando con el tiempo, así, a la anteriormente utilizada, establecida por el grupo Franco-Americano-Británico (FAB), basada en criterios morfológico-citoquímicos solamente, se le agregó la caracterización inmunofenotípica del Grupo Europeo para la Clasificación Inmunológica de Leucemias (EGIL) y finalmente se consideró también los aspectos citogenético y molecular que tienen aplicación pronóstica (1,7,18). La clasificación de la Organización Mundial de la Salud (OMS) se encargó de integrar e incluir todos estos aspectos, consensuándose los criterios de clasificación utilizados actualmente para las patologías oncohematológicas, que comprenden aspectos morfológico-citoquímicos, inmunofenotípicos, citogenéticos y moleculares (2,5,8). En base a estos criterios se estudiaron ambos pacientes constatándose que las células blásticas presentaron morfología, citoquímica e inmunohistoquímica confirmantes de origen mieloide con expresión de inmunofenotipo inmaduro. La detección de la translocación t(8;21) en los estudios citogenéticos fue importante para la sub-clasificación final completa, según estos criterios de la OMS y específicamente para determinar el factor de buen pronóstico en este tipo de LMA (13,14).
En cuanto al pronóstico de las leucemias en general, el estudio de las aberraciones cromosómicas, tanto numéricas como estructurales, constituye el mejor método para establecer el factor pronóstico en la actualidad (14, 15) y estas son detectables tanto por estudios citogenéticos convencionales (cariotipo), citogenética molecular (sondas fluorescentes de hibridización-FISH) u otras técnicas moleculares (PCR) (1, 3, 4, 6,19). Otro hallazgo citogenético que también es de interés pronóstico en LMA, es la pérdida del cromosoma Y en pacientes masculinos (8) lo cual hemos constatado en el caso clínico 2. Otros datos a tener en cuenta lo constituyen la edad, siendo peor el pronóstico en adultos mayores a 60 años que en el resto de la población y si se trata de una leucemia mieloide antes que linfoide o que esta sea secundaria antes que primaria (16). Cabe destacar aquí, que para el estudio completo de pacientes con enfermedad leucémica es importante seguir la secuencia correlacionada de criterios clínicos, morfológico-citoquímicos, inmunofenotípicos, citogenéticos y moleculares con la finalidad de llegar a una buena sub-clasificación diagnóstica y pronóstica en cuanto al tipo leucémico en cada caso. Esto permite al médico tratante emplear esquemas terapéuticos quimioterápicos ajustados a cada caso, consiguiendo la máxima eficacia de la medicación y aumentando la sobrevida libre de enfermedad en estos pacientes (2,20).
En nuestro estudio, en el caso clínico 1, no se realizó el tratamiento quimioterápico por falta del consentimiento de los padres, quienes retiraron al adolescente, que falleció en días posteriores siguiendo la evolución natural de la enfermedad. En este caso se destaca, cómo, la idiosincrasia propia de nuestro pueblo, especialmente de la población rural de escasos recursos, que ni ante la indicación ni ante los esfuerzos médicos continuos por instaurar el tratamiento correcto, teniendo buen pronóstico, muchas veces y probablemente ante la impotencia y la desesperación que acarrean los cuadros neoplásicos, no se convence y opta por la elección de tratamientos alternativos no exitosos en estos casos. Es lamentable que existan aún casos como este, en los que no se pueda instaurar la terapia específica, a pesar de todos los esfuerzos médicos.
El caso 2, fue sometido al régimen quimioterápico correspondiente con buena respuesta, consiguiéndose la remisión completa. Sin embargo, la aplasia medular e inmunosupresión subsecuentes a la fase de consolidación derivó en shock séptico que lastimosamente no pudo controlarse y el desenlace fue fatal para el paciente.
Con la descripción y el reporte de estos casos clínicos se pone en evidencia la relevancia que tienen los estudios secuenciales y correlacionados clínicos, morfológico-citoquímicos, inmunofenotípicos y citogenéticos que posibilitan la correcta sub-clasificación desde el punto de vista diagnóstico y pronóstico en los casos de patologías hematológicas como las leucemias agudas, teniendo en cuenta que aquellas de buen pronóstico tienen mucha chance de llegar a la remisión y curación.
REFERENCIAS BIBLIOGRÁFICAS
1. Beutler E, Lichtman MA, Coller BS, Kipps TJ, Seligsohn U. Hematología de Williams. 2a ed. Madrid: Marbán; 2007. [ Links ]
2. Kumar CC. Genetic abnormalities and challenges in the treatment of acute myeloid leukemia. Genes & Cancer. 2011;2(2):95-107. [ Links ]
3. Rodak BF. Hematología: fundamentos y aplicaciones clínicas. 2a ed. Buenos Aires: Médica Panamericana; 2004. [ Links ]
4. Sans-Sabrafen J, Besses Raebel C, Vives Corrons J. Hematología clínica. 4a ed. Madrid: Harcourt; 2001. [ Links ]
5. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H et al: WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: Internacional Agency for Research on Cancer; 2008. [ Links ]
6. Mufti GJ, Flandrin G, Schaefer HE, Sandberg AA, Kanfer EJ. An atlas of Malignant Haematology: Cytology, Histology and Cytogenetics. Martin Dunitz Ltd. London; 1996. [ Links ]
7. Kita K, Nakase K, Miwa H, Masuya M, Nishii K, Morita N,et al. Phenotypical characteristics of acute myelocytic leukemia associated with the t(8;21)(q22;q22) chromosomal abnormality: frequent expression of immature B-cell antigen CD19 together with the stem cell antigen CD34.Blood. 1992;80:470–7. [ Links ]
8. Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285-95. [ Links ]
9. Bakshi R, Zaidi SK, Pande S, Hassan MQ, Young DW, Montecino M et al. The leukemogenic t(8;21) fusion protein AML1-ETO controls rRNA genes and associates with nucleolar-organizing regions at mitotic chromosomes. J Cell Science. 2008;121:3981-90. [ Links ]
10. Elagib KE, Goldfarb AN. Oncogenic pathways of AML1-ETO in acute myeloid leukemia: multifaceted manipulation of marrow maturation. Cancer Lett. 2007; 251(2):179-86. [ Links ]
11. Blyth K, Cameron ER, Neil JC. The RUNX genes: gain or loss of function in cancer. Nature Rev. Cancer. 2005;5:376-87. [ Links ]
12. Schwieger M, Löhler J, Friel J, Scheller M, Horak I, Stocking C. AML1-ETO inhibits maturation of multiple lymphohematopoietic lineages and induces myeloblast transformation in synergy with ICSBP deficiency. J Exp Med. 2002;196:1227-40. [ Links ]
13. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nature Rev. Cancer. 2007;7:233-45. [ Links ]
14. Bullinger L,ValK PJ. Gene Expression Profiling in Acute Myeloid Leukemia. J Clin Oncol. 2005;23:6296-6305. [ Links ]
15. Martens JHA, Stunnenberg HG. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett. 2010;584:2662-9. [ Links ]
16. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics 2010. CA Cancer J Clin. 2010;60;277-300. [ Links ]
17. Figueredo SJ. Diagnóstico citohistoquímico sistemático en biopsias de médula ósea y citologías hematológicas en el Paraguay. TRABAJO DE TESIS. Facultad de Ciencias Médicas. Universidad Nacional de Asunción. Revista Anales de la Facultad de Ciencias Médicas-UNA. 2005;1-2(38):9-21. [ Links ]
18. European Group for the Immunological characterization of leukemias (EGIL): Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, et al. Proposals for the immunological classification of leukemias. Leukemia 1995;9:1783-6. [ Links ]
19. Da Silva G, Pilgerr D, De Castro S, Wagner S. Diagnostico laboratorial de leucemias mieloides agudas. Bras Patol Med Lab 2006.v42 p 77-84. [ Links ]
20. Fröhling S, Döhner H. Chromosomal abnormalities in cáncer. N Engl J Med 2008;359:722-34. [ Links ]
*Autor Correspondiente:Dra. Susy Figueredo Thiel, Dpto. de Patología. Instituto de Investigaciones en Ciencias de la Salud, Universidad Nacional de Asunción
Email: figuemed@hotmail.com Tel/Fax:021480185
Fecha de recepción: setiembre 2011; Fecha de Aceptación: Noviembre 2011.

