










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
versão On-line ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud v.9 n.1 Asunción jun. 2011
REPORTE DE CASOS
Diagnóstico prenatal del complejo agnatia holoprosencefalia
Prenatal diagnosis of the agnathia-holoprosencephaly complex
Oviedo P, *Ruoti Cosp M, Mendoza L, Ontano M, Irala L
Cátedra de Ginecología y Obstetricia. Facultad de Ciencias Médicas, UNA, Asunción, Paraguay
RESUMEN
El complejo agnatia holoprosencefalia o complejo disgnatia constituye un grupo de malformaciones severas que compromete el desarrollo del sistema nervioso central y de los arcos branquiales; casi siempre es incompatible con la vida y su extrema complejidad puede explicar su baja frecuencia, 1:100.000 neonatos.Primer caso reportado en la literatura paraguaya, gestante de 16 años, no exposición a teratógenos, remita al servicio. La ecografía informó polihidramnios, orbita ocular única, implantación baja de orejas bilateral y ausencia de cavidades bucal y nasal. Cariotipo 46XY. Nació por cesárea producto vivo, que fallece a los 15 minutos, presenta microcefalia, fontanelas cerradas, ciclopía, implantación baja de orejas, agenesia naso-bucal con esbozo único por debajo de la cavidad orbitaria con orificio permeable. Estudio radiológico: agenesia del maxilar inferior, con hipoplasia del maxilar superior. Autopsia: Holoprosencefalia alobar; ciclopía, sinoftalmia, sinotia, arhinia, agnatia; cardiopatía, pulmones hipoplásicos; criptorquidia bilateral; hipoplasia gástrica; y cordón umbilical con dos luces vasculares. La holoprosencefalia y ciclopía deben ser sospechadas durante la ecografía obstétrica de rutina y realizar ecografía detallada para corroborar diagnóstico y buscar otros defectos asociados. Es obligatoria la indicación de cariotipo fetal. Se recomienda el seguimiento obstétrico normal en los embarazos posteriores que continúan. No existen intervenciones fetales que cambien el pronóstico de los fetos con esta patología.
Palabras claves: Agnatia, holoproscencefalia, ciclopía, diagnóstico prenatal.
ABSTRACT
The agnathia-holoprosencephaly complex or disgnatia complex constitutes a group of severe malformations that compromises the development of the central nervous system and the branchial archs its low frequency, 1:100,000 neonates. This is the first case report in the Paraguayan literature, pregnant girl of 16 years old, no exposure to teratogens, remitted to the service. The ultrasound scan revealed polyhydramnios, single eye socket, low positioned ears and absence of oral and nasal cavities. Kariotype: 46XY. By C-section a baby was born alive but died at 15 minutes, presented microcefalia, closed fontanels, cyclopia, low positioned ears, oral-nasal agenesis with a single sketch below the eye socket with a permeable orifice. X-ray examination: agenesia del maxilar inferior, con hipoplasia del maxilar superior. Autopsy: alobar holoprosencephaly; cyclopia, synophtalmia, sinotia, arhinia, agnatia; cardiopathy, hypoplastic lungs; bilateral cryptorchidism; gastric hypoplasia and umbilical cord with two vessels. The holoprosencephaly and cyclopia should be suspected during the routine obstetric ultrasound and a detailed ultrasound should be performed to corroborate diagnosis and look for another associated defects. The indication of fetal karyotype is obligatory. The normal obstetric follow-up is recommended for the following pregnancies. There are not fetal interventions that change the prognosis of fetuses with this pathology.
Keywords: Agnatia, holoproscencephaly, cyclopia, prenatal diagnosis.
INTRODUCCIÓN
El complejo agnatia holoprosencefalia, es una malformación congénita mayor, casi siempre letal, que compromete el desarrollo del sistema nervioso central y de los arcos branquiales, caracterizada por ausencia o hipoplasia severa del maxilar inferior, posición anormal de los pabellones auriculares y holoprosencefalia (1).
La holoprosencefalia, rara entidad, consiste en una serie de anomalías congénitas cerebrales y del macizo facial, generadas en estadios muy tempranos del desarrollo, debida al fallo en la diferenciación y división del prosencéfalo, de manera que éste queda como una vesícula única incompletamente transformada en diencéfalo y telencéfalo con lóbulos y hemisferios. Esto causa defectos en el desarrollo de la línea media de la cara y en la estructura y la función del cerebro.
Tanto la complejidad como la severidad de las malformaciones pueden explicar su baja frecuencia, debido a que la mayoría de defectos de este tipo generan la interrupción temprana del embarazo. El Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC), indica una prevalencia de 1 en 100,000 casos(2) y alrededor de 100 casos se han comunicado en la literatura internacional (3).
Se presenta un caso deL complejo agnatia holoprosencefalia alobar asociado a ciclopía, sinoftalmia, sinotia, arhinia y se realiza una revisión de la literatura, en relación con la compleja etiología genética y embriológica de este conjunto de malformaciones mayores de la cara y el sistema nervioso central. Se trata del primer caso que se reporta en la literatura paraguaya.
CASO CLÍNICO
Paciente de 16 años de edad procedente de Ybycuí, con controles prenatales en el Hospital Distrital de la ciudad, controles prenatales sin registro, antecedentes personales, familiares y gineco-obstétricos sin datos de valor. Niega exposición a teratógenos. Se realizó ecografía en la semana 14 datos de interés. En la semana 24 informaron asimetría fetal (no especificada) y polihidramnios; y finalmente en la semana 27 importante polihidramnios, ausencia de cámara gástrica, microcefalia, no pudiéndose descartar hendidura palatina. Por esto fue remitida a nuestro servicio para mejor estudio con los diagnósticos de: 1. Nulípara gestante de 32 semanas, 2. Feto polimalformado y 3. Polihidramnios.
Al examen físico de ingreso se constató altura uterina de 46 cm, edema suprapúbico y de miembros inferiores hasta la rodilla.
Se realiza estudio ecográfico que informa polihidramnios severo, ciclopía (figura 1), microcefalia, ausencia de cámara gástrica, implantación baja de orejas bilateral y ausencia de cavidades bucal y nasal.
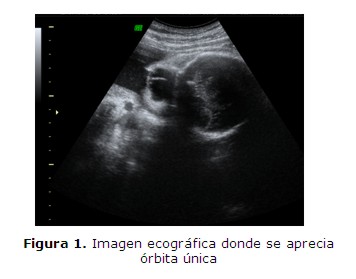
Al 6º día de internación se confirma por test de cristalización rotura prematura de membranas, escurriendo en escasa cantidad de líquido amniótico por vagina. Al 9º día de internación, se decide realizar la amnioreducción por compromiso del estado general (disnea y edema generalizado), extrayéndose 4.450 ml, se observa disminución de la altura uterina de 47 cm. a 37 cm. y un ILA de 63 cm. a 26 cm. con una mejoría en el estado general. Se constató bradicardia fetal previa y posterior al procedimiento. Se realizó además cordocentesis para estudio del cariotipo fetal, retornando 46, XY, sin alteraciones estructurales ni numéricas.
En el 17º día de internación se interrumpe la gestación por vía cesárea. Se obtiene un producto de sexo masculino de 34,7 semanas por Test de Capurro, 1550 g., Apgar 1/1, frecuencia cardíaca 60 lat/min y ausencia de respiración espontánea; polimalformado (figura 2 y 3): microcefalia con fontanelas cerradas, ciclopía (figura 4) (globos oculares fusionados en una única cavidad orbitaria), implantación baja de orejas, agenesia naso-bucal con esbozo único por debajo de la cavidad orbitaria (prosbócide) con orificio permeable que deja pasar la sonda 1 cm, criptorquidia izquierda y arteria umbilical única. No respiró espontáneamente, no habiéndose realizado estimulación ni maniobras de reanimación. Falleció a los 15 minutos.
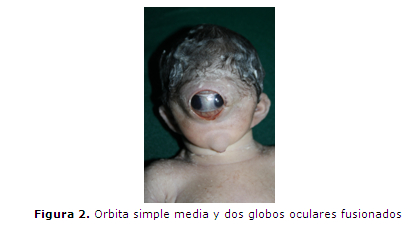
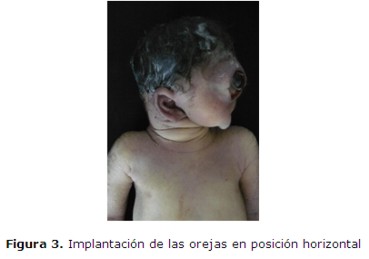
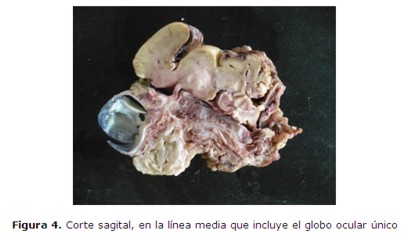
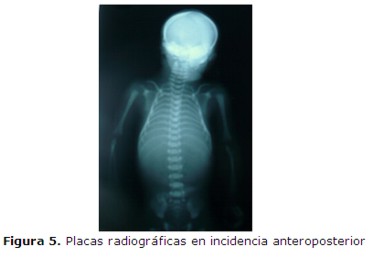
El estudio radiológico (figura 5) mostró: Agenesia del maxilar inferior (agnatia), con hipoplasia del maxilar superior; a nivel de calota impresiona aumento de tamaño de la fontanela anterior, calcificación ósea normal sin constatarse alteraciones de otras estructuras óseas.
La autopsia informó: Holoprosencefalia alobar; malformaciones faciales: ciclopía con sinoftalmia, sinotia, arhinia, agnatia; cardiopatía correspondiente a doble salida arterial del ventrículo derecho, con defecto del septo interventricular de localización subaórtica; pulmones hipoplásicos; criptorquidia bilateral; hipoplasia gástrica; placenta del tercer trimestre, membranas fetales sin alteraciones histológicas y cordón umbilical con dos luces vasculares.
DISCUSION
El complejo agnatia holoprosencefalia constituye un grupo de malformaciones severas que compromete el desarrollo del sistema nervioso central y de los arcos branquiales; el hecho de que casi siempre es incompatible con la vida por la obstrucción de las vías aéreas (4) y su extrema complejidad, pueden explicar su baja frecuencia.
La holoprosencefalia y ciclopía deben, ser sospechadas durante la ecografía obstétrica de rutina al hacer los cortes craneales sagitales para medir el diámetro biparietal, el perímetro cefálico y los cortes axiales de cara y cráneo. En estos casos se debe realizar una ecografía de detalle anatómico para corroborar el diagnóstico y buscar otros defectos asociados (5).
Cuando se descubre holoprosencefalia por examen ecográfico es obligatoria la indicación de cariotipo fetal. Se recomienda el seguimiento obstétrico normal en los embarazos posteriores. No existen intervenciones fetales que cambien el pronóstico de los fetos con esta patología.
La etiología es heterogénea. La mayor parte de los casos son esporádicos y de causa desconocida, pero se han informado casos de holoprosencefalia con un modo de herencia autosómica dominante, de penetrancia variable (6), autosómica recesiva (7) y recesiva ligada al cromosoma X (8).
Se propuso que las bases moleculares (9) de este trastorno eran las alteraciones en el gen Otx2 (orthodenticle drosophila homolog of 2), gen homeótico ubicado en el brazo largo del cromosoma 14q21-q22, que se expresa en las regiones dorsales y ventrales del telencéfalo, diencéfalo y mesencéfalo. Los efectos de las mutaciones del gen Otx2 y de sus genes modificadores, son la disminución de la capacidad inductiva del mesodermo precordial, que lleva a la agnatia por una migración neuronal anómala hacia las porciones ventrales del primer arco branquial y la segunda bolsa faríngea, fenómenos que suceden entre los 22 y 26 días de edad gestacional (10). La expresión del gen Otx2, quizá tenga relación con la vía de señalización Sonic Hedgehog, que es una familia de proteínas de señalización intercelular que juegan un papel primordial en el desarrollo embrionario; las mutaciones en esta vía generan diversos fenotipos que incluyen la holoprosencefalia lobar, alobar y semilobar, malformaciones que pueden cursar con cebocefalia, etmocefalia o probóscide (2).
En el caso que informamos, obviamente la inducción mesodérmica se vio severamente afectada en los arcos branquiales, Y por esta razón la implantación auricular fue en la línea media (sinotia), debido a la ausencia del maxilar inferior. Otro aspecto llamativo es la ciclopía, con la particularidad de ser un solo ojo en una sola orbita bien definida, lo que la diferencia de la ciclopía secundaria a la sinoftalmía o fusión ocular, donde las estructuras oculares y la bóveda orbital no están tan bien definidas. La ciclopía o sinoftalmía, son secundarias a la pérdida de estructuras mediales de la holoprosencefalia.
De tal modo que el defecto inicial pudo generarse en anomalías del desarrollo de las células que se derivan de las crestas neurales cefálicas por mutaciones en el gen Otx2, seguido de una inducción mesodérmica anormal en los arcos branquiales, ausencia del desarrollo en las estructuras de la línea media del cerebro secundario a falla en la estimulación del tubo neural y la consecuente ciclopía y probóscide, componentes del conjunto de anormalidades faciales de la holoprosencefalia.
Por otro lado, se puede asociar a alteraciones cromosómicas como la trisomía 13 (11). Se estima que más del 60% de los niños con trisomía 13 tienen holoprosencefalia; de modo recíproco, ésta se asocia a trisomía 13 en alrededor del 20% de los casos. Otras aneuploidías asociadas (12) incluyen las trisomías 18, 13q-, 18 q- y la triploidía (13).
El riesgo de recurrencia en ausencia de anomalías cromosómicas puede estimarse en un 6% (cifra que incluye tanto los casos verdaderamente esporádicos como los hereditarios, con un riesgo del 25-50%) (14).
REFERENCIAS BIBLIOGRAFICAS
1. Suárez-Obando F, Prieto JC. Complejo agnatia holoprosencefalia: informe de caso. Revista Colombia Médica 2007;38:305-7. [ Links ]
2. Martínez-Frías ML, Bermejo E. Frequency and trends of congenital defects in Spain: usefulness and significance of different frequencies. Med Clin (Barcelona) 1999;113:459-62. [ Links ]
3. Kauvar EF, Solomon BD, Curry CJR, Van Essen AJ, Janssen N, Dutra A, et al. Holoprosencephaly and agnathia spectrum: Presentation of two new patients and review of the literature. Am J Med Genet Part C Semin Med Genet 2010;154C:158–69. [ Links ]
4. Baker PA, Aftimos S, Anderson BJ. Case report. Airway management during an EXIT procedure for a fetus with dysgnathia complex. Pediatr Anesth 2004;14:781–6. [ Links ]
5. Wenghoefer M, Ettema AM, Sina F, Geipel A, Kuijpers-Jagtman AM, Hansmann H, et al. Prenatal ultrasound diagnosis in 51 cases of holoprosencephaly: craniofacial anatomy, associated malformations, and genetics. Cleft Palate Craniofac J 2010;47:15-21. [ Links ]
6. Erlich MS, Cunningham ML, Hudgins L. Transmission of the dysgnathia complex from mother to daughter. Am J Med Genet 2000; 95: 269-74. [ Links ]
7. Krassikoff N, Sekhon GS. Familial agnathia-holoprosencephaly caused by an inherited unbalanced translocation and not autosomal recessive inheritance. Am J Med Genet 1989; 34: 255-7. [ Links ]
8. Pauli RM, Pettersen JC, Arya S, Gilbert EF. Familial agnathia-holoprosencephaly. Am J Med Genet 1983; 14: 677-98. [ Links ]
9. Akagi T, Mandai M, Ooto S, Hirami Y, Osakada F, Takahashi M. Otx2 homeobox gene induces photoreceptor-specific phenotypes in cells derived from adult iris and ciliary tissue. Invest Ophthalmol Vis Sci 2004;45:4570-5. [ Links ]
10. Schiffer C, Tariverdian G, Schiesser M, Thomas MC, Sergi C. Agnathia-otocephaly complex: report of three cases with involvement of two different Carnegie stages. Am J Med Genet 2002;112:203-8. [ Links ]
11. Capobianco G, Cherchi PL, Ambrosini G, Cosmi E, Andrisani A, Dessole S. Alobar holoprosencephaly, mobile proboscis and trisomy 13 in a fetus with maternal gestational diabetes mellitus: a 2D ultrasound diagnosis and review of the literature. Arch Gynecol Obstet 2007;275:385-7. [ Links ]
12. Solomon BD, Rosenbaum KN, Meck JM, Muenke M. Holoprosencephaly due to numeric chromosome abnormalities. Am J Med Genet C Semin Med Genet 2010;154C(1):146-8. [ Links ]
13. Bekdache GN, Begam M, Al Safi W, Mirghani H. Prenatal diagnosis of triploidy associated with holoprosencephaly: a case report and review of the literature. Am J Perinatol 2009:479-83. [ Links ]
14. David AL, Gowda V, Turnbull C, Chitty LS. The risk of recurrence of holoprosencephaly in euploid fetuses. Obstet Gynecol 2007;110:658-62. [ Links ]
*Autor Correspondiente: Ruoti Cosp M. Cátedra de Ginecología y Obstetricia. Facultad de Ciencias Médicas, UNA. Asunción-Paraguay
Email:mruoticosp@hotmail.com. Fecha de Recepción: Abril de 2011, Fecha de aceptación: Mayo de 2011.

