










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
versão On-line ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud v.4 n.1 Asunción jun. 2006
REPORTE DE CASO
Monosomía 11 q compatible con síndrome de Jacobsen. Reporte de caso
11 q monosomy compatible with Jacobsen syndrome. Case report
*Torres E, Herreros MB, Rodríguez S, Ascurra M, Monjagata N
Departamento de Genética. Instituto de Investigaciones en Ciencias de la Salud (UNA) Asunción–Paraguay
RESUMEN
Se reporta el caso de una niña de 13 días de vida, con una deleción distal del brazo largo del cromosoma 11, nacida de madre portadora de un cromosoma marcador y de una inversión pericéntrica de la heterocromatina del cromosoma 9, la cual también se hallaba presente en la propósita. El cariotipo de la niña resultó 46,XX, del(11)(q24 –11qter), inv 9qh.La madre de la niña, con fenotipo normal, presentó un cariotipo 46,XX/47,XX+mar, inv9qh. El cariotipo del padre de la propósita fue normal. En este reporte destacamos la importancia de realizar el diagnóstico cromosómico en niños portadores de múltiples malformaciones y también la de efectuar el análisis cromosómico a los padres para el pronóstico del caso y asesoramiento genético de la pareja.
Palabras claves:Síndrome Jacobsen, deleción distal 11q, inversión 9qh.
ABSTRACT
This is the case of a 13-day girl with a distal deletion of the long arm of chromosome 11 and a pericentric inversion of the heterochromatin of chromosome 9. Her mother also had a pericentric inversion of the heterochromatin of chromosome 9 and a chromosome marker. The cariotype of the affected girl was 46, XX, del (11) (q24 –11qter), inv 9qh and the cariotype of the mother, with normal phenotype, was 46,XX,/47,XX+mar, inv 9qh. The mother of the proband had a normal phenotype. This paper highlights the importance of making an accurate chromosomal diagnosis in a child with multiple malformations as well as the importance of making a chromosomal analysis of the parents to make the case prognosis and proper genetic counselling.
Keywords: Jacobsen syndrome, 11q distal deletion, 9qh inversion.
INTRODUCCION
El desorden genético denominado Síndrome de Jacobsen (JBS) fue descrito por primera vez en 1973, asociado a una deleción distal del brazo largo del cromosoma 111–4. Durante el Quinto Taller Internacional sobre el mapa del cromosoma 11, en el año 1996, (Fifth Internacional Workshop on human chromosome 11 mapping 1996) se decidió denominar a la región 11q23–24.1, como locus del JBS5. La incidencia de este síndrome de microdeleción se estima en 1 de cada 100.000 recién nacidos1.
La deleción puede abarcar desde 11q23 hasta 11q25, pudiendo ser el origen citogenético de la misma, una segregación desbalanceada de progenitores portadores de una translocación recíproca balanceada o una segregación desbalanceada de novo, de progenitores normales. Otros rearreglos cromosómicos, como anillo del cromosoma 11, también han sido descritos asociados al Síndrome1,2,6.
Las deleciones terminales que involucran a la porción 11q23.3 suelen ser letales y no sobreviven por un tiempo mayor a las 35 semanas, a no ser que se hallen en mosaico con células normales1.
Hasta el año 1995 los signos clínicos descritos asociados al Síndrome de Jacobsen o monosomía parcial del cromosoma 11q fueron sutura metopica precoz y trigonocefalia, ptosis parpebral, hipertelorismo, boca en carpa, microretrognatia, orejas de implantación baja y malformadas así como anomalías cardíacas y retardo sicomotor3,7. Publicaciones posteriores describen nuevas manifestaciones fenotípicas, entre las que se incluye ano imperforado, catarata bilateral y pulmones hipoplásicos multilobulados8.
Se reporta el caso de una niña con Síndrome de Jacobsen, debido a una deleción distal del brazo largo del cromosoma 11, hija de madre portadora de un cromosoma marcador y de una inversión pericéntrica de la heterocromatina del cromosoma 9.
REPORTE DEL CASO
Paciente de sexo femenino, producto del primer embarazo, padre de 28 y madre de 26 años respectivamente, no consanguíneos. Prenatal controlado, con valores de HCG negativos hasta las 5 semanas de embarazo y luego positivo, pero con valores muy por debajo de lo normal para la edad gestacional (6–7 semanas). Durante el embarazo la madre refiere permanentes cefaleas y dolores tipo premenstruales, con leve hemorragia hasta los tres meses. Por ecografía, se detectó la presencia del signo del limón. Nace a las 36 semanas, con un peso de 2.700 g, percentil 50; circunferencia cefálica de 33,5 cm, percentil 25 y una talla de 47 cm, percentil 25. No precisó incubadora, sí oxígeno por algunas horas, siendo intervenida a las siete horas de nacida debido a ano anterior imperforado. La historia familiar refiere que un primo de la madre tuvo un niño con ano imperforado.
La niña es derivada a los diez días de vida a la consulta de genética clínica debido a sus características fenotípicas: sutura metópica prominente; ojos con inclinación mongoloide, hipertelorismo, puente nasal alto, orejas pequeñas de implantación baja y hélices dismórficos; micrognatia, cuello corto; soplo cardíaco; ano anterior operado por imperforación; dedos largos, pliegue palmar único bilateral y pliegue único en ambos quintos dedos. (figuras 1 y 2)
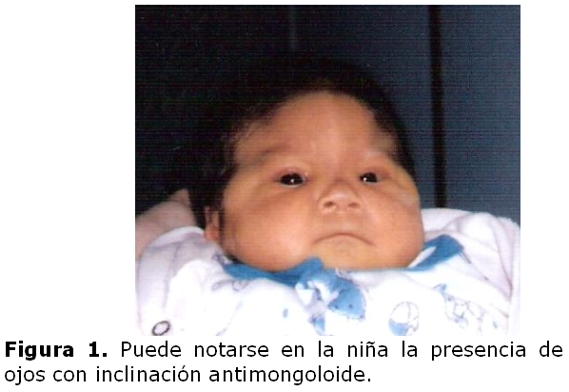
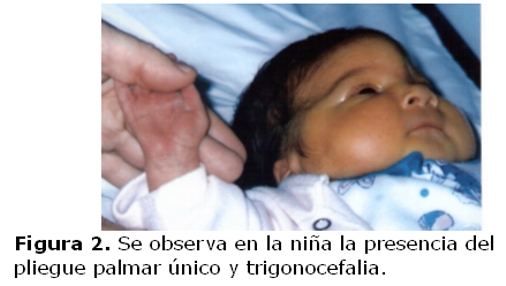
Por ecocardiografía se detectó comunicación interauricular, tipo fosa oval de 3mm, e insuficiencia leve de la válvula tricúspide, la radiografía de cráneo reveló trigonocefalia, el estudio por TAC de cráneo asimismo detectó fusión prematura de sutura metópica y la radiografía de tórax resultó normal. Fallece a los 11 meses por complicaciones de una neumonía. La madre con fenotipo normal y sin antecedentes de aborto.
Se estudiaron cromosómicamente la niña y sus progenitores. El diagnóstico cromosómico se llevó a cabo a través del cultivo de linfocitos de sangre periférica, en medio RPMI enriquecido con 15% de suero fetal bovino y fitohemoaglutinina, los que se cultivaron a 37ºC durante 72 horas. Transcurrido este tiempo se agregó colchicina (0,003ug/ml) durante 90 minutos, posterior tratamiento con solución hipotónica de KCl (0,075M), y se fijaron en solución (3:1) de metanol:ácido acético. Se realizaron los extendidos sobre portaobjetos, se tiñeron con Giemsa para el estudio convencional y el análisis cromosómico se realizó con técnicas de bandas G y de bandas C.
Se analizaron citogenéticamente cincuenta células de la niña. Se observó en todas la metafases analizadas la presencia de una deleción en la región distal del brazo largo de uno de los cromosomas del par 11 correspondiente a la región q24.1. Así como también una inversión pericéntrica de la heterocromatina de uno de los cromosomas del par 9. El cariotipo del padre fue normal y en la madre se observó en el 5% de las metafases analizadas, la presencia de un cromosoma marcador y además la inversión del cromosoma 9.
El cariotipo de la niña presentó 46,XX, del(11)(q24–qter),inv9qh. (figura 3)
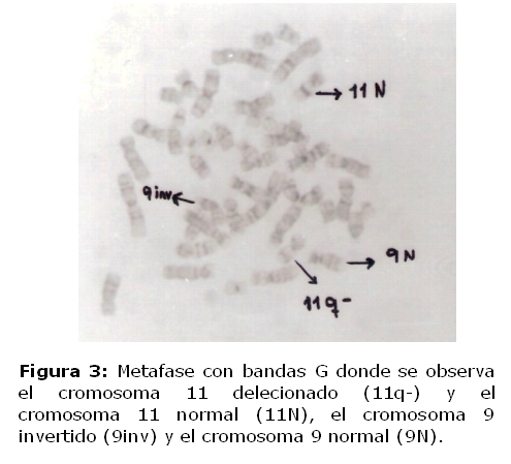
El cariotipo de la madre reveló 46,XX,/47,XX+mar,inv9qh y el cariotipo del padre fue 46,XY.
DISCUSION
El Síndrome de Jacobsen es reconocido desde el año 1996, como una microdeleción de la región 11q23–24.1, ya sea hereditario o de novo5. No obstante, el cromosoma 11 puede presentar otras deleciones en su brazo largo, la más común es la porción 11q23–11qter, también se han reportado casos de deleciones intersticiales de la región 11q23–11q25 en individuos con fenotipos típicos de este síndrome. Aparentemente la ausencia de la banda 11q24.1 es la que corresponde a la región crítica que determina la aparición de este síndrome. Aproximadamente el 85% de los casos son de novo7. El análisis cromosómico realizado en la paciente presentó una deleción en la región 11q24–qter, con lo cual se confirmó el Síndrome de Jacobsen en la misma.
Para la determinación del origen de la anomalía, hereditaria ó de novo, y habiéndose realizado los estudios cromosómicos a los padres, estos evidenciaron un cariotipo normal en el padre, pero en la madre se detectó una inversión pericéntrica de la heterocromatina del cromosoma 9 y la presencia de un cromosoma marcador en el 5% de las células analizadas. Si bien, las inversiones pericéntricas del cromosoma 9 involucrando la región heterocromática son rearreglos relativamente comunes en la población general, es decir, son consideradas variantes normales en la población general, existen reportes de familias que portan esta alteración y se transmiten de generación en generación, incluso asociados a diversos fenotipos, como esterilidad, infertilidad, aborto habitual y en raros casos con otras alteraciones cromosómicas; su importancia en el análisis de las poblaciones humanas no ha sido aún lo suficientemente explorada9-12. Entonces, si consideramos que la presencia de esta inversión no estaría en relación con las malformaciones presentes en la niña, nuestro análisis se orienta entonces en el cromosoma marcador, el cual consiste en un pequeño fragmento cromosómico. Para determinar su origen y para realizar un asesoramiento genético adecuado, es importante su identificación, pero debido a que las técnicas citogenéticas convencionales en general no permiten identificarlo, se debe realizar en la madre el estudio cromosómico con técnicas citogenéticas moleculares, como la Hibridación in situ con sondas fluorescentes (FISH). Aún así podemos inferir que dicho cromosoma marcador podría haberse originado de una translocación, lo que a su vez estaría originando por una segregación meiótica desbalanceada, el Síndrome de Jacobsen en la niña.
BIBLIOGRAFIA
1. Penny LA, Dell´Aquila M, Jones MC, Bergoffen J, Cunniff C, Fryns JP et al. Clinical and molecular characterization of patients with Distal 11q Deletions. Am J Hum Genet 1995; 56:676–83. [ Links ]
2. Hustinx R, Verloes A, Grattagliano B, Herens C, Jamar M, Soyeur D et al . Monosomy 11q: report of two familial cases and review of the literature. Am J Med Genet 1993; 47:312–7. [ Links ]
3. Lewanda AF, Morsey S, Reid ChS, Jabs EW. Two Craniosynostotic patients with 11q deletions, and review of 48 cases. Am J Med Genet 1995; 59:193–8. [ Links ]
4.Neavel CB, Soukup S. Deletion of (11) (q24.2) in a mother and daughter with similar phenotypes. Am J Med Genet 1994; 53:321–4. [ Links ]
5. Shows TB, Alders M, Bennett S, Burbee D, Cartwright P, Chandrasekharappa S. Report of the Fifth International Workshop on human Chromosome 11 Mapping 1996. Cytogenet Cell Genet 1996; 74 (1–2):1–56. [ Links ]
6.Fryns JP, Kleczkowska A, Buttiens M, Marien P, Van Den Berghe H. Distal 11q monosomy. The tipical 11q monosomy syndrome is due to deletion of subband 11q24.1. Clinical Genetics 1986; 30: 255–60. [ Links ]
7. Donnenfeld A, Zackai E, Emanuel B. Chromosome 11, monosomy 11q. En: Buyse ML, ed.Birth Defects Encyclopedia Dover, MA: Center for Birth Defects Information Services; 1990. p. 360–1. [ Links ]
8. Puvabanditsin S, Garrow E, Zia-Ullah MO,Lianthanasarn P, Denev KI. Monoxomy 11Q: Report of new phenotypic manifestations. Genet Couns 2001; 12(3): 283–6. [ Links ]
9. Velázquez M, Tavara C, Manya W, Costa D. Heterocromatina Constitutiva en una población escolar. Resultados preliminares. En: 30a Congreso Nacional de Genética Humana; 2005; San Luis Potosí, México; Asociación Mexicana de Genética Humana, A.C. (Edición Especial Número 5). [ Links ]
10. Margarit E, Soler A, Carrió A, Costa D, Ballesta F. Genética Básica (Y II): Citogenética. Jano 2001; 61(1403): 41–50. [ Links ]
11. Pérez–Caballero–Macarrón C, Quintana Castilla A, Aparicio Mies JM. Blefarofimosis, ptosis, epicanto inverso y telecanto asociado a sordera Neurosensorial. An Esp Pediatr 1999; 51:530–2. [ Links ]
12. Levy B, Jalal S, Dunn T, Warburton P, Tonk V, Hirschhorn K, et al. Unique case of Mosaicism Involving two morphologically similar Marker Chromosomes of Different Centric Origin in a patient with Developmental Delay. Am J Med Genet 2002; 108:198–204. [ Links ]
*Autor Correspondiente: Lic. Elodia Torres
Dpto. de Genetica, Inst. Inves. Ciencias de la Salud
Río de la Plata y Lagerenza. CC:2511. Asunción–Paraguay
E–mail:genetica@iics.una.py

