










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
versão On-line ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud v.1 n.1 Asunción 2002
REPORTE DE CASO
Displasias Oseas: A Proposito de cuatro nosologias diferentes
Skeletal Dysplasias: About four different Nosologies
*Ascurra M, Herreros MB y Rodríguez S
Departamento de Genética, Instituto de Investigaciones en Ciencias de la Salud, Universidad Nacional de Asunción
RESUMEN
Las displasias esqueléticas son patologías que presentan una alteración generalizada del tejido óseo. Las mismas constituyen una de las causas más frecuentes de retardo severo del crecimiento y si bien algunas son más comunes que otras, en conjunto presentan una elevada frecuencia de aparición. Según la porción ósea afectada pueden ser rizomélicas (porción proximal: brazo y muslos), mesomélicas (porción media: antebrazo y pierna) y acromélicas (porción distal: mano y pie) o combinaciones de los diferentes tipos. En este trabajo se presentan: el diagnóstico diferencial, pronóstico, posibilidades de diagnóstico prenatal y asesoramiento genético, de cuatro tipos de displasias óseas. Los mismos corresponden a pacientes evaluados en el Departamento de Genética del Instituto de Investigaciones en Ciencias de la Salud, tres del sexo masculino y una del femenino, con un rango de edad comprendido entre 3 días y 21 meses de edad. Para el diagnóstico diferencial se utilizaron los datos clínicos, el examen radiográfico y el árbol genealógico, identificándose una hipoacondroplasia, displasia tanatofórica, displasia diastrófica y una acondroplasia. Este último caso corresponde al hijo de una madre afectada por la misma patología. Se resalta la importancia del diagnóstico y diferenciación precoces para el pronóstico y determinación del origen, teniendo en cuenta que algunas displasias óseas son letales y que en las hereditarias existe el riesgo de repetición.
Palabras claves: Displasias óseas, Retardo del crecimiento, Condrodisplasias, Condodistrofias.
ABSTRACT
Skeletal dysplasias are pathologies which present a general alteration of the osseus tissue and they constitute one of the most common causes of severe growth deficiency. Some of them are more common than others but they all have a high frequency of appearance. According to the affected osseus portion, they may be rhizomelic (proximal portion: arms and thighs), mesomelic (media portion: forearms and legs), acromelic (distal portion: hands and feet) or a combination of the different types. The differential diagnosis, prognosis, possibilities of prenatal diagnosis and genetic counselling of four types of skeletal dysplasias are presented. The four patients, three boys and a girl, were seen at the Genetics Department of the IICS and their ages ranged from 3 days to 21 months. The differential diagnosis was based on clinical features, radiographic studies and genealogical tree. We identified one case each of diastrophic dysplasia, hypochondroplasia, diastrophic and achondroplasia. The mother of this last case was affected by the same pathology. We would like to highlight the importance of an early diagnosis and differentiation in the prognosis and determination of the etiology, considering that some osseus dysplasias are lethal and that hereditary forms have a repetition risk.
Keywords: Skeletal dysplasias, Growth deficiency, Chondrodysplasias, Chondrodistrophies.
INTRODUCCION
Las displasias esqueléticas (DE) son patologías que presentan una alteración generalizada del tejido óseo y constituyen una de las causas más frecuentes del retardo severo del crecimiento. Si bien algunas son más comunes que otras, en conjunto presentan una frecuencia de aparición elevada de aproximadamente 1 en 3.000 a 5.000 recién nacidos. Por ejemplo la frecuencia para la displasia tanatofórica es de 1 por cada 20.000 recién nacidos, la acondroplasia 1 por cada 10.000 a 15.000, la hipocondroplasia 1/12 de la frecuencia estimada para la acondroplasia y si bien la frecuencia de la displasia diastrófica no esta aún determinada, se considera una displasia ósea bastante común (1, 2, 3). Hasta hace poco tiempo el diagnóstico preciso de una DE era solo perinatal, a través de la clínica, la bioquímica y la radiología. El advenimiento de las técnicas de Biología Molecular ha permitido el diagnóstico prenatal de muchas de ellas, permitiendo la adopción de medidas y el consejo genético correspondiente (4). El diagnóstico y diferenciación precisos de una DE permiten otorgar un pronóstico, teniendo en cuenta que algunas de ellas son letales, y un asesoramiento genético, ya que según el tipo de DE, el patrón de herencia será Autosómico Recesivo (AR) o Autosómico Dominante (AD). En este último grupo, las mutaciones frescas ocurren en más del 80% de los casos, siendo la edad paterna avanzada (50 años en adelante) un factor predisponente para ello (5).
Caso clínico Nro. 1
Niño de un año y cinco meses al momento de la consulta, con talla de 70 cm (perc. 3) y perímetro cefálico de 52 cm (+2DS), nacido a los ocho meses de gestación por cesárea, con un peso de 2.800 gr. Primer hijo de un padre de 25 años, sano y una madre de 24 años de edad. La madre es acondroplásica al igual que la hermana gemela de la abuela materna (ver árbol genealógico, Figura 1). Al examen físico el niño presentó: macrocefalia, braquidactilia, frente prominente, hipoplasia mediofacial, miembros cortos, lordosis lumbar, manos con dedos cortos y signo del tridente. A los 8 y 10 meses presentó otitis media sin perforación del tímpano y aún no se sentaba por lo que fue referido a fisioterapia. Las imágenes radiológicas del niño revelaron un angostamiento caudal del canal de la espina dorsal con pedículos cortos y metacarpos y falanges cortas en mano. Por las características clínico-radiológicas, se realiza el diagnóstico de acondroplasia.
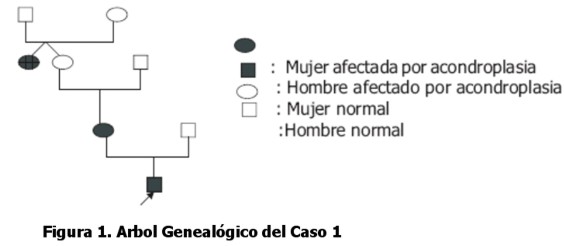
Caso clínico Nro. 2
Niño de 1 año y 9 meses, talla de 72 cm ( p3), peso: 10 kg (perc. 3) y circunferencia cefálica de 50 cm (perc. 50) al momento de la consulta, nacido en un parto normal, con un peso de 2.550 gr., talla de 44 cm y circunferencia cefálica de 34 cm. Primer hijo de un padre de 41 años y 1,72 cm de estatura, en quien llaman la atención los miembros cortos y el tronco largo. Madre de 34 años y 1,47 cm de estatura. No hay antecedentes patológicos maternos en relación al embarazo, ingestión de medicamentos o consanguinidad. Al examen físico presentó: frente abombada, implantación baja de orejas, orificio preauricular derecho, clinodactilia bilateral de ambos quintos dedos, genu varo, 2º ortejo mal posicionado y corto. La radiografía mostró huesos tubulares cortos con ligero ensanchamiento en las metafisis. Cuello del fémur ancho y corto. Peroné distal largo, cúbito distal corto y estiloides cubital largo. Actualmente el niño tiene 4 años y su desarrollo psicomotor es normal. Por las características clínico-radiológicas se realiza el diagnóstico de hipocondroplasia.
Caso clínico Nro. 3
Recién nacido del sexo femenino, de 34 semanas por Capurro, talla de 36 cm, perímetro cefálico 35 cm y peso de 2.400 gr. Nacida por cesárea, por presentación cefálica y con diagnóstico probable de acondroplasia. Padres aparentemente normales, no consanguíneos. Madre de 33 años, 3 gestaciones, un aborto espontáneo de 8 semanas; sin antecedentes de patologías o ingestión de medicamentos durante el embarazo. Al examen físico la niña presentó: macrocefalia, frente amplia, puente nasal chato, proptosis, orejas de implantación baja. Cuello corto, tórax estrecho, miembros cortos con dedos cortos en forma de salchicha, pliegue palmar único bilateral. Las radiografías revelan escápulas cuadradas, huesos de la pelvis pequeños, costillas cortas, columna con espacios intervertebrales aumentados, se observa platiespondilia y vertebras en forma de U. Fémures y húmeros cortos y arqueados, estos últimos con forma de tubo de teléfono. Ecografía abdominal con hidronefrosis bilateral. Debido a los problemas respiratorios que presentó desde el nacimiento, se la internó en terapia intensiva y se la conectó un respirador. A los 7 días, falleció por insuficiencia respiratoria. Por las características clínico¬radiológicas, se realizó el diagnóstico de displasia tanatofórica.
Caso clínico Nro. 4
La niña nacida por cesárea en presentación pelviana y con circular de cordón, con talla de 38 cm, muy por debajo del percentil 3, peso de 2.850 gr. (perc. 3) y circunferencia cefálica de 36 cm. (perc.75). Queda internada por tres días debido a dificultad respiratoria. Producto del primer embarazo de una madre de 25 y padre de 28 años. No hay antecedentes patológicos del embarazo, de ingestión de medicamentos, ni antecedentes familiares. La niña tenía una talla de 40 cm (perc.3) y un perímetro cefalico de 39,5 cm (perc. 75) y las siguientes características clínicas: Hipoplasia medio facial, protopsis ocular, puente nasal chato, base ancha, filtrum corto y narinas antevertidas, un angioma plano en dorso nasal, filtrum, entrecejas y frente. Orejas quísticas. Tórax estrecho, miembros superiores cortos y luxación de ambos codos. Mano con pliegue único bilateral, pulgares en abducción, otros dedos con desviación cubital. Miembros inferiores cortos, talipes equinovarus 1º ortejo en posición vara, amplia diastasis entre primero y segundo ortejos. A los 3 meses tenía una talla de 45 cm (perc.3) y circunferencia cefálica de 41 cm ( perc. 98). A los 11 mese s peso de 7 kg. (perc. 3), talla de 53 cm (perc. 3) y circunferencia cefálica de 46 cm (perc. 50). Desarrollo psicomotor normal. Radiográficamente se observan huesos tubulares anchos y cortos con desarrollo de metáfisis anchas y epífisis aplanadas e irregulares. Platispondilia. Primer metacarpo muy corto con el pulgar proximal. Por las características clínico-radiológicas, se realiza el diagnóstico de Displasia Diastrófica.
DISCUSION
En ocasiones, la malformación o el cuadro que presentan las DE es tan específico que puede hacerse el diagnóstico con relativa facilidad. Un ejemplo de ello son las orejas quísticas en la displasia diastrófica. Otros puntos presentados son el asesoramiento genético, el diagnóstico prenatal ecográfico y la posibilidad de estudio por biología molecular (3, 6, 7). En cuanto a los casos presentados el primero corresponde a una acondroplasia, con patrón autosómico dominante (riesgo genético 50%), penetrancia completa (de estar presente el gen, éste se manifiesta), por lo que la recurrencia familiar solo es posible explicar en nuestro caso a través de la existencia, ya sea de un mosaicismo germinal o una premutación inestable presente en la gemela normal (8). El segundo caso corresponde a una hipoacondroplasia, autosómica dominante, pero con expresividad variable pudiendo pasar desapercibida y por lo tanto no ser diagnosticada, sin que se lleve cabo el asesoramiento genético hasta la presentación de individuos severamente afectados, ya sea por el retardo o la talla. En todas las DE, el primer y más importante paso es el de determinar si la misma es o no letal. Esta información no solo es importante en lo que respecta a lo que se dirá a los padres, sino en cuanto a las medidas terapéuticas a ser tomadas, ya que estas serían inútiles en la displasia tanatofórica, como en el 3er caso, donde la muerte se produce irremediablemente por la hipoplasia pulmonar existente. El cuarto caso presentado corresponde a una displasia diastrófica, con patrón de herencia autosómica recesiva y por tanto un riesgo del 25% para la descendencia futura. En cuanto al tratamiento en las DE, éste es muy variable y depende de la patología específica. En la acondroplasia y la hipocondroplasia, la hormona de crecimiento se está probando todavía con resultados dispares. El más utilizado es el alargamiento óseo que si bien es largo y difícil para el paciente y los familiares, en general tiene un resultado final satisfactorio, especialmente en la hipoacondroplasia. En resumen, es de extrema importancia realizar un diagnóstico prenatal preciso, aunque teniendo en cuenta el número y variedad de DE existentes, suele ser difícil conocer el fenotipo y los rasgos característicos de cada una de ellas. El diagnóstico de letalidad es el que condiciona el manejo de una gestación y, una vez que ésta termina y se confirma la displasia, el tratamiento y asesoramiento genético para las futuras generaciones.
BIBLIOGRAFÍA
1. Andersen PE. Prevalence of lethal osteochondrosdysplasias in Denmark. Am J Med Genet 1989; 32:484. [ Links ]
2. McKusik Va. On Line Mendelian Inheritance in Man. USA: John Hopkins University Press; 1996. [ Links ]
3.Jones KL. Smith`s Recognizable Patterns of Human Malformation. 5ta edición. USA: WB Saunders Company; 1997. [ Links ] Cap 7.
4. Seino Y, Moriwake T, Tanaka H, Inoue M, Kanzaki S, Tanaka T, Matsuo N, Niimi H. Molecular defects in achondroplasia and the effects of growth hormone treatment. Ac Pediatr Suppl 1999; 428:118-20. [ Links ]
5.Orioli IM, Castilla E, Scareno G, Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia and osteogenesis imperfect. Am J Med Genet 1995; 59:209-217. [ Links ]
6. Gorlin RJ, Cohen MM, Levin Jr. S: Syndromes of the Head and the Neck. 3ra. Ed. USA: Oxford University Press; 1990. [ Links ]
7. Rimoin DL, Connor JM, Pyeritz R, E. Emery and Rimoin`s Principles and Practice of Medical Genetics. 3ra. Ed. USA: Edit. Churchill Livingstone; 1997. [ Links ]
8.Dondival P, Le Marec B. Genetic couseling in unexpected familial recurrence of achondroplasia. Am J Med Genet 1987; 28:949-954. [ Links ]
*Autor Correspondiente:Dra. Marta Ascurra, Departamento de Genética Instituto de Investigaciones en Ciencias de la Salud. Río de la Plata y Lagerenza. Asunción-Paraguay
Email: genetica@iics.una.py


{kind=link}