










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMemorias del Instituto de Investigaciones en Ciencias de la Salud
versão On-line ISSN 1812-9528
Mem. Inst. Investig. Cienc. Salud v.1 n.1 Asunción 2002
REPORTE DE CASO
Craniosinostosis: Revisión y Presentación de Casos
Craniosinostosys: Revision and Case Reports
*Herreros MB, Ascurra M
Departamento de Genética, Instituto de Investigaciones en Ciencias de la Salud, Universidad Nacional de Asunción
RESUMEN
La craniosinostosis consiste en la fusión prematura de las suturas del cráneo y junto con otras anomalías, constituye un síndrome. Son patologías de etiología y patogenia heterogénea. Se conocen más de 90 de síndromes de craniosinostosis sin incluir las craniosinostosis ocasionales, ni las secundarias. El diagnóstico es clínico-radiólógico y la prevalencia al nacimiento de todos los tipos de craniosinostosis es de 343 por un millón. La sinostosis de la sutura sagital es la más frecuente seguida por la coronal. La fusión prematura de las suturas metópica y lambdoidea es menos frecuente y los casos de craniosinostosis aislada son esporádicos. En el caso de las sindromáticas, pueden ser mutaciones frescas o de transmisión autosómica dominante. En este grupo las anomalías asociadas más frecuentes son las de miembros pero también se ven malformaciones cardiacas, genitourinarias y defectos de oído. En algunos casos, se observa retraso mental. La aparición de craniosinostosis en la descendencia se encuentra relacionada con edad paterna avanzada. En este trabajo, presentamos 6 casos de craniosinostosis distribuidos de la siguiente forma: dos varones con síndrome de Apert, un ñiño y una niña con síndromes de Pfeiffer (uno de ellos con cráneo en trébol), un niño con síndrome de Jackson Weiss y una niña con craniosinostosis aislada (cráneo en trébol). Se hará una revisión de las craniosinostosis más frecuentes insistiendo en los síndromes presentados. Se presentan estos casos por la importancia del diagnóstico preciso de los mismos, ya que de ello dependen el tratamiento y manejo de los pacientes así como la realización de un Asesoramiento Genético sobre las posibles complicaciones como: retraso mental, malformaciones cardiacas (dependiendo del síndrome) y del riesgo de recurrencia en la familia.
Palabras claves:Craniosinostosis, Sutura, Mutación, Síndrome.
ABSTRACT
Craniosynostosis is a disorder of the skull shape caused by a premature fusion of the sutures. Together with other anomalies, it constitutes a syndrome. Craniosynostosis is a pathology of heterogenic etiology. There are more than 90 syndromes of craniosynostosis known, excluding the occasional and secondary craniosynostosis. The diagnosis is made clinically and radiologically and the prevalence of all types is 343 per million of newborns. The most frequent synostosis is the fusion of the sagital suture followed by the fusion of the coronal. The premature fusion of the metopic and lambdoid sutures is less frequent. Cases of isolated craniosynostosis are sporadic. The syndromatic craniosynostosis may be fresh mutations or of autosomal dominant inheritance. In the group of syndromatic craniosynostosis, the more frequently associated anomalies are those of the limbs though cardiac malformations, genitourinary anomalies and ear defects are also seen. In some cases, there is also mental retardation. Craniosynostosis in the offspring is related to father's old age. In this paper, we present six cases of craniosynostosis: two boys with Apert syndrome, a boy and a girl with Pfeiffer syndromesl (one of them with cloverleaf skull), one boy with Jackson Weiss syndrome and one girl with isolated craniosynostosis (cloverleaf skull). We will make a revision of the more frequent syndromes of craniosynostosis emphazising the syndromes of our current cases. We report these cases because of the importance of a precise diagnosis, since patient handling and treatment depend on it. It is also important in order to provide proper genetic counseling about the possible complications such as mental retardation, cardiac malformations and others (depending on the syndrome) and to inform the risk of recurrence in the family.
Keywords: Craniosynostosis, Suture, Mutation, Syndrome.
INTRODUCCION
La craniosinostosis consiste en la fusión prematura de las suturas del cráneo, de etiología y patogenia heterogénea, con una prevalencia al nacimiento de todos los tipos de 343 por un millón y diagnóstico clínico radiológico. La fusión de la sutura sagital es la más frecuente, seguida de la coronal, metópica y lambdoidea. La forma de la cabeza depende de la sutura que esté fusionada y de l momento durante el desarrollo del feto en que se produjo la unión. Las craneosinostosis se pueden presentar en forma aislada o asociadas a otras anomalías constituyéndose así en un síndrome. Se conocen más de 90 síndromes de craniosinostosis sin incluir los ocasionales y los de origen secundario (1). Los casos de craniosinostosis aislada son esporádicos. Las sindromáticas tienen un patrón de herencia autosómico dominante, la mayoría de penetrancia completa y con una alta frecuencia de mutaciones frescas relacionadas con la edad paterna avanzada (2). En el caso de la craniosinostosis familiar, puede estar afectada la misma o diferentes suturas en los distintos miembros de la familia. El cráneo en trébol constituye una variedad de craneosinostosis, en la que el cráneo adopta la forma de un trébol de tres hojas, siendo los síndromes de presentación más frecuentes el síndrome de Pfeiffer tipo 2 y la Displasia Tanatofórica tipo 2 (3,4). Las craniosinostosis sindromáticas se suelen presentar asociadas a otras malformaciones, las más frecuentes son las de los miembros pero también se ven malformaciones cardiacas, genitourinarias y defectos de oído. El riesgo de retraso mental se encuentra presente siempre que haya una craniosinostosis, independientemente de la etiología. Se sabe que ciertos síndromes lo presentan con mayor frecuencia, como el síndrome de Apert y otros, raramente, como el síndrome de Crouzon (2, 5) Este a su vez puede estar presente en síndromes como el de Saethre Chotzen, en el que el retraso mental es considerado raro, lo que fue descrito por Bianchi y col., 1985 (6). El retraso mental es más frecuente en el caso de fusión bilateral de las suturas coronales que de la sagital y es 2,5 veces más frecuente cuando la fusión es bilateral que unilateral de cualquier sutura. Entre los síndromes de craniosinostosis probablemente el más grave sea el de Apert, ya que además de la craniosinostosis, retraso mental y sindactilia blanda y/o dura de manos y pies, también hay una afectación importante de boca y paladar, generalmente con paladar hendido, úvula bífida y con mala oclusión severa (7). La prevalencia del síndrome de Apert es de aproximadamente 15,5 por millón de nacimientos, igual a la del síndrome de Crouzon (8). Por otro lado, existen casos de craniosinostosis asociadas tanto a efectos ambientales como a anomalías cromosómicas. La identificación cromosómica permitió el mapeo y la individualización de la mutación molecular de algunos síndromes de craniosinostosis como los síndromes de Apert, Crouzon, Pfeiffer, Jackson Weiss y también para las craniosinostosis tipo Boston. Existen trabajos que relacionan la aparición de craniosinostosis en los hijos de padres que trabajan en agricultura, forestación y mecánica. En este grupo, se observó más riesgo en los recién nacidos de sexo masculino. No se reporta una asociación importante entre craniosinostosis y la ocupación materna (8).
PRESENTACION DE LOS CASOS
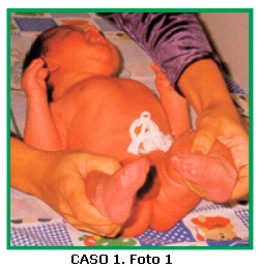
CASO 1. Foto 1 Paciente de sexo masculino, de 6 días de edad, que consulta por dismorfias faciales, braquiturricefalia, paladar hendido y sindactilia de manos y pies. El niño es el producto del primer embarazo de una pareja joven, no consanguínea. No hay antecedentes familiares, patológicos, ni de ingestión de medicamentos durante el embarazo. El padre es comerciante. Al examen físico: talla: 51,5 cm (p50) peso: 3.600 gr (p75) CC:35,5 cm (p50).
CC- circunferencia cefálica.
Cabeza: braquiturricefalia, diástasis de frontales y parietales.
Ojos: proptosis, inclinación antimongoloidea de fisuras palpebrales.
Nariz: puente nasal alto, nariz prominente y larga, punta bulbosa.
Boca: normal, paladar hendido (duro y blando) Orejas: implantación baja.
Manos: ambas con sindactilia de 2° a 5° dedos, pulgares anchos y con desviación radial, pliegues palmares únicos bilaterales.
Pies: sindactilia de 2° a 5° ortejos, ambos halluces en posición vara.
Genitales normales, no se auscultan soplos.
Ecocardiografía: normal. Ecografía renal: normal.
Radiografía de cráneo: revela fusión de suturas coronales y lambdoidea.
Diagnóstico clínico radiológico: Síndrome de Apert. El niño falleció a los 4 meses de síndrome de muerte súbita del lactante.
CASO 2. Foto 2
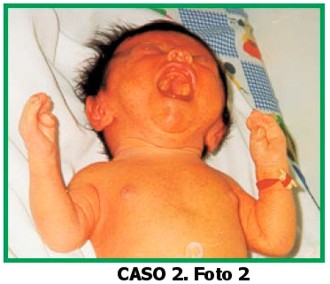
Paciente de sexo masculino, de 2 días de edad, que consulta por dismorfias faciales, braquiturricefalia, paladar hendido y sindactilia de manos y pies. El niño es el producto del sexto embarazo de una madre de 39 años y padre de 43 años, no consanguíneos.
No hay antecedentes amiliares, no hay antecedentes patológicos del embarazo actual, ni de ingestión de medicamentos.
La madre tuvo un aborto espontáneo. El padre refiere haber estado expuesto a insecticidas por 25 años, es agricultor. Al examen físico: talla: 53 cm (p75) peso: 3.300 gr (p 50) CC:34,5 cm (p50).
Cabeza: braquiturricefalia, diástasis de frontales y parietales.
Ojos: proptosis, inclinación antimongoloidea de fisuras palpebrales. Hemangioma plano en frente.
Nariz: puente nasal alto, nariz prominente y larga, punta hacia abajo. Boca: normal, paladar hendido (duro y blando).
Orejas: implantación baja.
Manos: ambas con sindactilia de 2° a 5° dedos, pulgares anchos y con desviación radial. Pliegues palmares únicos bilaterales.
Pies: sindactilia de todos los ortejos. Halluces en posición vara. Genitales normales, no se auscultan soplos.
Ecocardiografía: normal.
Ecografía renal: normal. Radiografía de cráneo: revela fusión de suturas coronales y lambdoidea.
Diagnóstico clínico radiológico: Síndrome de Apert. El niño se encuentra en buen estado de salud, a los 6 meses del diagnóstico.
CASO 3.Foto 3
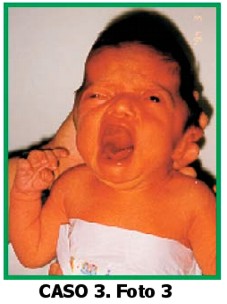
Paciente de sexo femenino, de 9 días de edad, que consulta por dismorfias faciales, cráneo en trébol y pulgares anchos. La niña es el producto del quinto embarazo de una pareja joven, no consanguínea. No hay antecedentes familiares, no hay antecedentes patológicos del embarazo actual, ni de ingestión de medicamentos. El padre es comerciante. Al examen físico: talla: 52 cm (p75) peso: 3.600 gr (p50).
CC: 31,5 cm (-2DS). Cabeza: cráneo en trébol, diástasis de frontales y parietales.
Hipoplasia mediofacial.
Ojos: proptosis, inclinación antimongoloidea de fisuras palpebrales.
Nariz: en pico de loro. Boca: normal, paladar normal.
Manos: ambas con pulgares anchos y desviación radial.
Orejas:implantación muy baja.
Pies: ambos halluces anchos.
Genitales normales, no se auscultan soplos.
Ecocardiografía: normal.
Ecografía renal: normal. Radiografía de cráneo: revela fusión de suturas coronales y lambdoidea, cráneo en trébol. Diagnóstico clínico radiológico: Síndrome de Pfeiffer tipo 2. La niña fue intervenida quirúrgicamente para liberar las suturas fusionadas. Se le instaló una válvula para derivación de hidrocefalia, se encuentra en recuperación.
CASO 4. Foto4A, 4B
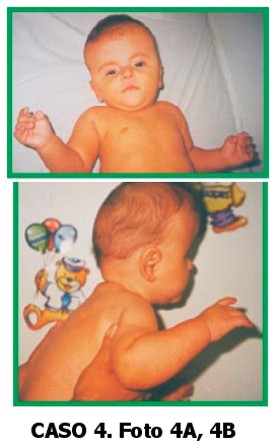
Paciente de sexo masculino, de 6 meses de edad, que consulta por braquiturricefalia y sindactilia de 2° y 3° ortejos de ambos pies. El niño es el producto del sexto embarazo de una madre de 35 años, segundo hijo con el actual padre de 32 años.
Sin antecedentes de consanguinidad, ni patológicos o de ingestión de medicamentos durante el embarazo. El padre es comerciante. La madre tiene manos y pies con pulgares y halluces anchos, compatible con síndrome de Pfeiffer y refiere que de pequeña tenía la cabeza deformada.
Al examen físico: talla: 68 cm (p25) peso: 7.900 gr (p25) CC: 42 cm (p10) Cabeza: braquiturricefalia, fontanela anterior cerrada, suturas coronales sobreelevadas.
Ojos: inclinación antimongoloidea de fisuras palpebrales.
Nariz: puente nasal chato.
Boca: normal, paladar alto. Hipoplasia maxilar.
Orejas:oreja izq. implantación baja; derecha, normal.
Manos: pulgares cortos, anchos y con desviación radial.
Los dos quintos dedos en clinodactilia y campodactilia de ambos índices y pulgares.
Pliegues palmares normales. Pies: halluces anchos y en posición vara.
Sindactilia parcial entre 2° y 3° ortejos.
Genitales normales, no se auscultan soplos.
Ecocardiografía: normal Ecografía renal: normal.
Diagnóstico clínico radiológico: Síndrome de Pfeiffer tipo 1.
El niño está en buen estado de salud y no tiene retraso del desarrollo sicomotor.
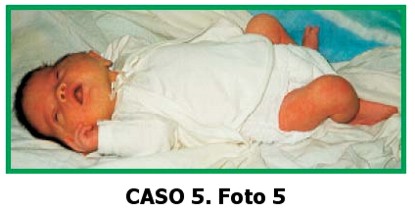
CASO 5. Foto 5
Paciente de sexo femenino de 2 días de edad, que consulta por dismorfias faciales y cráneo en trébol. La niña es el producto del quinto embarazo de una madre de 33 años y padre de 41, no consanguíneos. Embarazo sin antecedentes de patologías, ni de ingestión de medicamentos. La madre refiere una niña fallecida a los tres meses por una cardiopatía congénita y un mortinato, de los tres embarazos con este padre. El padre es comerciante.
Al examen físico: talla: 49 cm (p50) peso: 3.200 gr (p50) CC:35,5 cm (p50)
Cabeza:cráneo en trébol. Hipoplasia mediofacial.
Ojos: sin proptosis.
Nariz: puente nasal alto, narinas antevertidas. Boca: normal, paladar normal.
Orejas: implantación baja.
Manos: normales. Pies: normales.
Genitales normales, no se auscultan soplos.
Ecocardiografía: normal.
Ecografía renal: normal.
Electroencefalograma: Discretos signos de disfunción de estructuras subcorticales de carácter inespecífico, con predominancia en regiones frontales.
Estudio cromosómico: normal. Radiografía de cráneo: cráneo en trébol, fusión de suturas coronales y lambdoidea.Diagnóstico clínico radiológico: Cráneo en trébol aparentemente esporádico. La niña se encuentra en buen estado de salud.
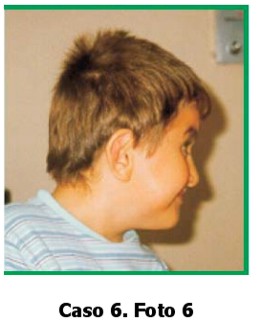
CASO 6. Foto 6
Paciente de sexo masculino, de 6 años de edad, que consulta por retraso mental moderado, fenotipo sindromático, braquicefalia y y halluces en posición vara. El niño es el producto del cuarto embarazo de una pareja joven, no consanguínea.
No hay antecedentes familiares, patológicos, ni de ingestión de medicamentos durante el embarazo. El padre es empleado de un comercio. Al examen físico: talla: 120 cm (p75) peso: 23 kg (p75) CC: 50 cm (p25).
Cabeza: braquicefalia. Ojos: normales. Nariz: puente nasal alto, nariz prominente y larga, punta hacia abajo. Boca: normal, paladar normal. Hipoplasia maxilar.
Orejas: implantación baja. Manos: dedos cortos. Pliegue palmar único bilateral.
Pies: ortejos cortos, ambos halluces en posición vara. Genitales normales, no se auscultan soplos. Ecocardiografía: normal Ecografía renal: normal.
Radiografía de cráneo: revela fusión de suturas coronales, lambdoidea y sagital.
Radiografía de pies: fusión del primero y segundo metatarsos, fusión de los huesos navicular y cuneiforme.
Diagnóstico clínico radiológico: Síndrome de Jackson Weiss El niño se encuentra en buen estado de salud y presenta un retraso mental moderado.
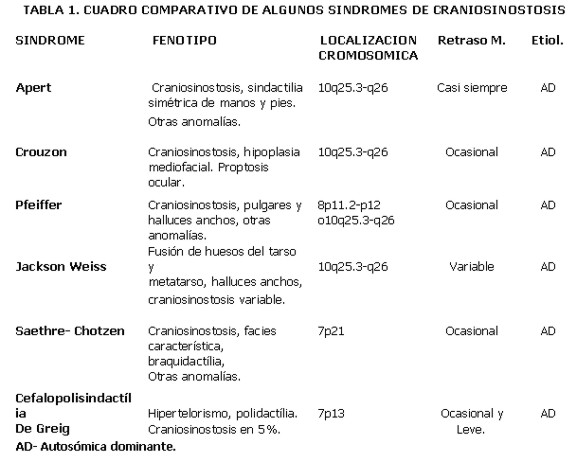
A continuación incluimos un cuadro comparativo de las craniosinostosis más frecuentes, su fenotipo, localización cromosómica, presencia o ausencia de retraso mental y etiología. El diagnóstico diferencial de estos síndromes es muy importante para realizar el asesoramiento genético.Tabla 1.
DISCUSION
Estas craneosinostosis se presentan por la importancia de realizar un diagnóstico preciso y asesoramiento genético, ya que la mayoría de las craniosinostosis sindromáticas son autosómicas dominantes, con un riesgo del 50% de recurrencia para la descendencia de los afectados. En el caso de las craniosinostosis esporádicas es indispensable descartar una patología cromosómica. Entre los casos presentados, los casos 1 y 2 son síndromes de Apert considerados mutaciones frescas, ya que los padres son sanos. El caso 3 es un síndrome de Pfeiffer tipo 2, también considerado mutación fresca, con padres sanos. El caso 4 es un síndrome de Pfeiffer tipo 1, heredado de la madre, con un riesgo de repetición del 50% para los futuros hijos. El caso 5 es un cráneo en trébol esporádico y se descartó la probable etiología cromosómica. El caso 6 es un síndrome de Jackson Weiss, considerado mutación fresca, con padres sanos. El tratamiento consiste en la liberación quirúrgica temprana de las suturas y el concepto actual es que si bien mejora la forma de la cabeza probablemente no actúe sobre el retraso mental. También se deben tratar las anomalías asociadas, como las cardiacas, sindactilia de pies y manos o cualquiera que pueda encontrarse y se debe hacer un acompañamiento cercano del niño para ver su desarrollo intelectual. En los casos de mutaciones frescas es realmente muy difícil la prevención ya que no existen causas específicas de estos síndromes. En los casos heredados, la forma de prevenirlos es realizando el Asesoramiento Genético Familiar.
BIBLIOGRAFIA
1. Gorlin RJ. Cohen M, Levin LS. Syndromes of the head and the neck. 3º Edición. USA: Oxford University Press; 1990; Cap. 14-15. [ Links ]
2. Jones KL. Smiths recognizable patterns of human malformation. 5º Edición. USA: W.B. Saunders Company; 1997; Pag.412-431. [ Links ]
3. Moore Qs, Banik S. Ultrasound scanning in a case of thanatophoric dwarfism with cloverleaf skull.Br J of Radiology 1980; 53:241. [ Links ]
4. Chervenak FA, Blakemore KJ, Isaacson G, Mayden K, Hobbins JC. Antenatal Sonographic Findings of Thanatophoric Dysplasia with Cloverleaf Skull. Am J Obstet Gynecol 1983;146:984-5. [ Links ]
5. Murdoch-Kinch CA, Bixler D, Ward RE. Cephalometric Analysis of Families with Dominantly Inherited Crouzon Syndrome: An aid to Diagnosis in Family Studies. American Journal of Medical Genetics 1998; 77:405-411. [ Links ]
6. Bianchi E, Arico M, Podestá AF, Grana M, Fiori P, Beluffi G. A family with the Saethre Chotzen Syndrome. Am J Med Gen 1985; 22:649-658. [ Links ]
7. Bradley CM, Alderman BW, Williams MA, Chekoway H, Fernbach SK, Greene C, Bigelow PL, Reif JS. Parental Occupations as Risk Factors for Craniosynostosis in Offspring. Epydemiology 1995; Vol 6 (3): 306-310. [ Links ]
8. Taybi H, Lachman RS. Radiology of syndromes, metabolic disorders and skeletal dysplasias. 4° Edición. USA: Editorial Mosby; 1996. [ Links ]
*Autor Correspondiente: Dra. Mara Herreros, Departamento de Genética Instituto de Investigaciones en Ciencias de la Salud. Río de la Plata y Lagerenza. Asunción-Paraguay
Email:genetica@iics.una.py

