











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCION
La Atrofia Muscular Espinal (AME) engloba una serie de enfermedades heterogéneas caracterizadas por degeneración y pérdida de las neuronas motoras de la médula espinal y del tronco encefálico. Se divide en 2 grupos según el origen genético: AME 5q ligado al gen de supervivencia de la motoneurona 1 (gen SMN1), la forma más frecuente, 95% y AME no 5q ligado a otros genes1.
La AME 5q es debida a mutaciones en el gen SMN1 que provoca degeneración y pérdida neuronal motora con la consiguiente debilidad y atrofia muscular progresiva.1 Es una de las principales causas genéticas de muerte en niños. Se hereda con un patrón autosómico recesivo. Se estima a nivel mundial: una prevalencia de 1-2:100.000 personas, una incidencia de 1:6.000-10.000 nacimientos y una frecuencia de portadores de 1: 40-70 individuos2,3.
Todos los pacientes con AME 5q presentan como signos clínicos: debilidad generalizada de predominio proximal, reflejos osteotendinosos disminuidos o ausentes, compromiso de músculos respiratorios (intercostales, relativa preservación del diafragma) y nivel cognitivo conservado4. La AME presenta un espectro clínico de presentación variable, que va desde una manifestación muy severa de inicio en los primeros meses de vida a una forma muy leve de inicio en la adulta. Se la clasifica en 4 tipos según la edad de inicio de los síntomas y el máximo logro motor alcanzado5,6.
La confirmación diagnóstica se realiza por estudio genético-molecular debido a que todos los tipos de AME 5q presentan ausencia o mutación bialélica en el gen SMN1. Estudios complementarios como enzimas musculares, pruebas electrofisiológicas y biopsia muscular ayudan a distinguirla de otras enfermedades neuromusculares7.
Durante la evolución de la enfermedad aparecen complicaciones de carácter progresivo: respiratorias, nutricionales, osteoarticulares secundarias a la debilidad muscular. Las respiratorias son las principales complicaciones y son causa de morbimortalidad sobre todo en pacientes con AME tipo I, con menor severidad y frecuencia en los tipos II y III 8,9.
Las intervenciones terapéuticas deben realizarse precozmente, para evitar o disminuir las complicaciones, y debe incluir la participación de la familia y de un equipo de salud multidisciplinario. Existen guías actualizadas de cuidados estándar para pacientes con AME, que aplicadas junto con las terapias modificadoras específicas para AME 5 q, desarrolladas en los últimos años, han cambiado la evolución natural de la enfermedad. Estas terapias actúan ya sea reemplazando el gen SMN1 o modulando el ARN del gen SMN2, y produciendo así el incremento de la proteína SMN completa y funcionante esencial para la supervivencia de las neuronas motoras y el desarrollo de hitos motores nunca alcanzados10,11.
El objetivo del estudio fue describir las características clínicas, epidemiológicas y genéticas de los pacientes pediátricos con AME 5q que fueron evaluados en el servicio de Neurología del Hospital General Pediátrico Niños de Acosta Ñu.
MATERIALES Y MÉTODOS
Diseño y lugar del estudio: Estudio observacional, descriptivo, retrospectivo, de corte transversal, llevado a cabo en el Hospital General Pediátrico Niños de Acosta Ñu, San Lorenzo, Paraguay. Población y reclutamiento: por medio de un muestreo no probabilístico de casos consecutivos, fueron incluidos en el estudio Pacientes con diagnóstico de AME evaluados en el servicio de Neurología del Hospital General Pediátrico Niños de Acosta Ñu en el periodo comprendido desde julio del 2013 hasta julio del 2017. Se revisaron las historias clínicas de los pacientes y en los casos en que no se encontraban todos los datos se realizaba una comunicación telefónica para completar lo faltante.
Variables estudiadas: Sexo, Procedencia, Motivo de consulta, Manifestaciones clínicas, Clasificación de AME, Edad de diagnóstico clínico, Edad de diagnóstico genético, Mutación genética, Métodos auxiliares de diagnóstico, Antecedentes familiares, Evolución, Tratamiento, Mortalidad, Edad de fallecimiento.
Tamaño de la muestra: por tratarse de una enfermedad poco frecuente se presenta una serie de casos.
Procesamiento de los datos y plan de análisis: los datos fueron analizados en el sistema SPSSv21 utilizando estadística descriptiva. Las variables cualitativas se expresaron en porcentajes y las cuantitativas en medias con desviación estándar o medianas de acuerdo a su distribución. Se consideró un error alfa menor a 5%. El protocolo fue aprobado por el comité de investigación y ética institucional con liberación de consentimiento informado por tratarse de un estudio retrospectivo.
RESULTADOS
Fueron incluidos 26 pacientes con diagnóstico de AME que fueron evaluados en el servicio de Neurología del Hospital General Pediátrico Niños de Acosta Ñu de julio de 2013 a julio del 2017. El predominio fue de sexo masculino 15/26 (57,5%). La mayoría provenía del departamento central 10/26 (38,5%) y capital 3/26 (11,5%). En cuanto al motivo de consulta de los pacientes, se describen en la Tabla 1.
En relación a la clínica presentada por los pacientes en el momento de la consulta, la misma se detalla en la Tabla 2.
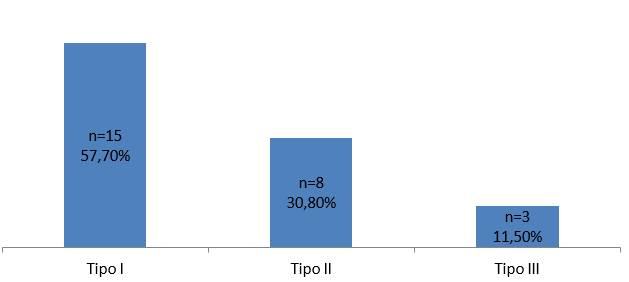
La frecuencia de clasificación de los tipos de AME encontradas en los pacientes se observa en la Figura 1.
La mediana de edad de diagnóstico clínico fue de 6 meses, mínimo 2 meses y máximo 48 meses, con intervalos intercuartiles de P25: 4 meses y P75: 20,2 meses.
La mediana de edad de diagnóstico genético fue de 7,5 meses, mínimo 3 meses y máximo 51 meses.
Sólo 14/26 pacientes se realizaron estudio genético molecular. Todos los pacientes que se realizaron el estudio genético molecular presentaron la mutación genética típica: delección homocigota de los exones 7 y 8 del gen SMN1.
En cuanto a los métodos auxiliares de diagnóstico realizados a los pacientes, los mismos se muestran en la Tabla 3.
Solo 4/26 (15,4%) tenían antecedentes familiares y/o sintomatología relacionada con AME. En 2 familias se encontraron hermanos con AME (1hermano en cada familia) y en otras 2 familias se encontraron hermanos con debilidad leve-moderada en miembros inferiores (1 hermano en cada familia). No se encontró consanguinidad.
Con respecto a las complicaciones presentadas se encontraron: Defunción 12/26(46,2%), Mixta (más de una complicación: respiratorio, articular, nutricional) 9/26(34,6%), Alteraciones osteoarticulares 4/26 (15,4%) y ninguna 1/26 (3,8%).
El tratamiento recibido por los pacientes incluía soporte respiratorio, nutricional, rehabilitación motora, ortopédico.
La mortalidad fue del 46,1% (12/26). Todos los fallecidos fueron pacientes con AME tipo I. La mediana de edad de fallecimiento fue de 8 meses, mínimo 3 meses y máximo 2
Tabla 1 Motivos de consulta de los pacientes con diagnóstico de AME que consultaron en el servicio de Neurología del HGP. N=26
| Motivo de consulta | N | % |
| No camina | 5 | 19,2 |
| Dificultad respiratoria y escaso movimiento | 5 | 19,2 |
| Escaso movimiento | 4 | 15,4 |
| Dificultad respiratoria | 4 | 15,4 |
| Dificultad p/ pararse | 3 | 11,5 |
| No sostén cefálico y escaso movimiento | 1 | 3,8 |
| No sostén cefálico | 1 | 3,8 |
| Dificultad p/ subir escalera | 1 | 3,8 |
| Caídas frecuentes y dificultad p/ subir escalera | 1 | 3,8 |
| Caídas frecuentes | 1 | 3,8 |
| Total | 26 | 100 |
Tabla 2 Manifestaciones Clínicas en la exploración física de los pacientes con diagnóstico de AME que consultaron en el servicio de Neurología del HGP. N=26
| Manifestación clínica | N | % |
| Fasciculación lingual, hipotonía, arreflexia | 15 | 57,7 |
| Poliminimioclonus, hipotonía, arreflexia, escoliosis | 5 | 19,2 |
| Hipotonía, arreflexia, escoliosis | 3 | 11,5 |
| Fasciculación lingual, hipotonía, arreflexia, artrogriposis | 1 | 3,8 |
| Fasciculación lingual, hipotonía, arreflexia, escoliosis | 1 | 3,8 |
| Fasciculación lingual, hipotonía, arreflexia, poliminimioclonus, escoliosis | 1 | 3,8 |
| Total | 26 | 100 |
Tabla 3 Métodos auxiliares de diagnóstico realizados a los pacientes con diagnóstico de AME. N=26
| Método auxiliar | N | % |
| CPK, Aldolasa, ENG(*),EMG (**) | 19 | 73,1 |
| CPK, Aldolasa | 7 | 26,9 |
| Total | 26 | 100 |
**)ENG: Electroneurografía, (**) EMG: Electromiografía
DISCUSIÓN
En éste trabajo la mayoría de los pacientes con AME consultaron por debilidad, hipotonía, dificultad respiratoria y retraso en el desarrollo motor; en el momento de la evaluación física predominaron la fasciculación lingual, la hipotonía y la arreflexia para el tipo I, y el poliminimioclonus y la debilidad muscular para los tipos II y III, coincidiendo con resultados encontrados en un estudio multicéntrico realizado en Chile, con el mismo tamaño de muestra, en un período de 7 años, así en éste estudio chileno todos los tipo I (4/26) consultaron por hipotonía y dificultad respiratoria y presentaron fasciculación lingual, hipotonía y arreflexia al momento de la consulta, todos los casos con AME tipo II (11/26) y AME tipo III (11/26) consultaron por algún grado de retraso motor y físicamente todos presentaron debilidad muscular (22/26) mientras que poliminimioclonus 8/11 para el tipo II y 10/11 para el tipo III12. En éste estudio hubo predominio del sexo masculino, datos similares se han reportado en un estudio retrospectivo realizado en España, que incluyó 37 pacientes, evaluados en un período de 25 años13.
En esta serie, la AME tipo I representó más de la mitad de los casos, siendo el tipo más frecuente, similar a los reportado en la bibliografía, seguido del tipo II y por último el III. Un estudio de 105 pacientes con diagnóstico de AME realizado en Cuba, entre 1997 y 2011, encontró que 59% de los casos presentaba AME tipo I; 28,6% AME tipo II y 12,4% AME tipo III14. La mediana de edad de diagnóstico clínico fue tardío para las expectativas de mejoría clínica que las medidas terapéuticas tempranas prometen.
Las concentraciones de CPK en pacientes con AME tipo I suelen ser normales y en pacientes tipo II y III ligeramente elevados, que coincide con los resultados de los pacientes del presente trabajo. La cuantificación de CPK permite descartar miopatías. La electroneurografía muestra un patrón de denervación axonal aunque también puede reportar un resultado normal. A la electromiografía reporta un patrón neurogénico de denervación. Castiglioni et al. reportaron signos de denervación y reinervación crónica con potenciales de unidad motora de amplitud y duración acrecentadas y reclutamiento disminuido en pacientes con AME I y II, en cambio en pacientes con AME III reportaron signos de reinervación crónica con potenciales de unidad motora de gran amplitud. En esta serie se realizaron a 19/26 pacientes con resultados que coinciden con lo mencionado. Los estudios electrofisiológicos siguen siendo de apoyo para el diagnóstico de AME, sobre todo en casos atípicos. En este estudio a ningún paciente se le realizó biopsia de músculo y nervio 15,16.
Éste estudio ejemplifica el amplio espectro clínico de esta enfermedad poniendo de manifiesto la mayor dificultad diagnóstica en aquellos casos menos graves clasificados como AME tipo III y confundiendo con otras enfermedades neuromusculares, principalmente las miopatías. Esto hace que sea necesaria la realización de estudios específicos y menos invasivos como el genético molecular. Nicole S y coautores recuerdan publicaciones de la era pre genética de la enfermedad en que se describía que cerca de un cuarto de los pacientes con diagnóstico clínico de AME tipo III se presentaban con fenotipo de miopatía aumento de los niveles sanguíneos de CPK e histopatología miopática17.
La alteración genética más frecuente en éste trabajo fue la delección en homocigosis de los exones 7 y 8. En Cuba, Carlos Viñas P. y coautores en su estudio de 105 pacientes con AME encontraron un 36,2% de pacientes con delección homocigota de los exones 7 y 8; un 35,2% con delección homocigota del exón 7 solamente, y un 28,2% con otro tipo de mutación14. No se realizó la cuantificación de copias del gen SMN2 en ningún paciente. En todos los pacientes que no pudieron realizarse el estudio genético molecular, la clínica y los métodos auxiliares fueron determinantes para el diagnóstico.
El antecedente de familiares con AME es un dato de alerta para el médico de atención primaria por lo que debe de interrogarse siempre en el niño con debilidad y/o hipotonía. En España, Madrid Rodríguez y su grupo reportan 62% de antecedentes familiares13. En Colombia, en cambio, este dato coexiste en 13,7% de los pacientes con AME18. En esta serie se encontraron 4 casos con antecedentes de familiares y/o sintomatología relacionada con AME.
A la fecha actual no existe tratamiento curativo para esta enfermedad. Hasta hace pocos años el tratamiento ha sido sólo de soporte o paliativo, centrados principalmente en los cuidados de los problemas neuromusculares, respiratorios, nutricionales y digestivos. Se elaboraron guías específicas de cuidados, con una visión integral, sistematizada y multidisciplinaria de los diferentes tipos de AME y de las complicaciones que surgen con la evolución de la enfermedad19,20.
La mayoría de los pacientes de esta serie recibió fisioterapias motora y respiratoria más soporte nutricional (en especial los pacientes con AME I y II), soportes básicos recomendados por las guías internacionales de cuidados de la atrofia muscular espinal. Ninguno de los pacientes evaluados recibió terapia específica modificadora. El tratamiento sigue siendo sólo medidas de apoyo conforme a lo establecido en las guías de trabajo actualizadas en el 201819,20.
Consta en este estudio el fallecimiento de 15/15 pacientes con AME tipo I, siendo la mediana de edad de fallecimiento menor de un año, lo cual está en un rango inferior a lo publicado hasta ahora por la incorporación tardía al soporte respiratorio activo y nutricional. En un estudio español fueron seguidos 25 pacientes con AME, la mayoría presentaba AME tipo I (18 casos), de éstos 17 fallecieron, siendo la mediana de supervivencia en esa serie para el tipo I de 8 meses13.
CONCLUSIÓN
Los motivos de consulta más frecuentes fueron la debilidad, la hipotonía, la dificultad respiratoria y el retraso en el desarrollo motor. La AME tipo I fue la forma clínica más frecuente. La edad de diagnóstico clínico y genético fue tardía. La mortalidad encontrada fue elevada, y sólo presente en los pacientes con AME tipo I.
En el estudio realizado se encontró un patrón clínico, epidemiológico y genético clásico con diagnóstico relativamente tardío.

