









 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
La Enfermedad de Gorham-Stout, también denominada la enfermedad del hueso fantasma, u osteólisis masiva idiopática, es un trastorno óseo poco común caracterizado por una osteólisis progresiva con proliferación linfática y vascular. La incidencia es muy baja, con solo unos pocos cientos de informes de casos descritos en la literatura. El trastorno no parece mostrar preferencia por raza, área geográfica o sexo, aunque puede ser ligeramente más prevalente en los hombres, la mayoría de los casos que se han descrito que han ocurrido antes de los 40 años, con una edad promedio de 25 años1.
Fue descrita inicialmente por Jackson en 1838 y posteriormente clasificado por Gorham y Stout en 1955. A nivel mundial, considerando todos los potenciales afectados sólo se han notificado 200 casos. La osteólisis puede afectar cualquier hueso, más frecuentemente el tórax, el fémur, mandíbula, pelvis, escápula, húmero, vértebra, tibia, clavícula, y articulaciones. La fisiopatogenia exacta de esta entidad aún no ha sido completamente aclarado. Varios estudios han demostrado que factor de crecimiento endotelio vascular (VEGF-A) e interleucina-6 puede estar elevado en la circulación periférica de los individuos afectados, así como otros biomarcadores2.
La detección de marcadores inmunohistoquímicos de los canales linfáticos sugiere que la malformación linfática es la patología primaria. Sin embargo, la etiología todavía es especulativa sin vínculo hereditario, neoplásico o infeccioso y alrededor del 50% de todos los pacientes presentan un episodio de trauma antes del diagnóstico3.
La pérdida ósea se produce debido al reemplazo de tejido graso intramedular normal y médula ósea con vasos linfáticos proliferantes4. El curso clínico varía ampliamente, desde remisión espontánea de la osteólisis progresiva con algunas leves manifestaciones clínicas a un curso clínico fatal. Se ha descrito una mortalidad media del 13%. Los hallazgos histopatológicos incluyen linfáticos y tejido vascular en el hueso e inmunohistoquímica D2-40, tiene una sensibilidad del 92,6% y especificidad del 98,8%.
El tratamiento no está definido. Las opciones siguen siendo un desafío y a menudo se satisfacen específicamente al caso individual.
Se presenta un caso que significó un desafío diagnóstico en el hospital General Pediátrico “Niños de Acosta Ñu”.
CASO CLINICO
Paciente escolar de 5 años de edad, de sexo femenino, ingresa al HGP por tercera vez, remitida de un Sanatorio privado del interior del País con el diagnostico de Quilotórax espontáneo de etiología a determinar + Tumor óseo de fémur izquierdo.
Antecedentes previos a la remisión: Presenta una historia pre hospitalaria de fiebre (39 grados) y dificultad respiratoria importante de 24hs de evolución que requirió ingreso hospitalario. Se realizó radiografía de tórax, mostró velamiento de campo pulmonar derecho. Se realizó punción - drenaje de 600 ml de líquido lechoso y se instaló tubo de drenaje pelural. Recibió Cefotaxima por 9 días y Vancomicina por 7 días, posteriormente se remite a nuestro Hospital para estudio y tratamiento.
APP: Paciente con antecedentes de dificultad para la marcha desde los 2 años de edad, tratada con medicación sintomática. En el año 2018 por persistencia se síntomas se realiza radiografía de miembro inferior izquierdo, que revelaba una disminución de la densidad ósea; se realizó biopsia de la lesión, que mostró: Tejido osteocartilaginoso cortical y trabecular osteomedular benigno y sin alteraciones microscópicas significativas. Fué remitida a un centro privado de la capital para colocación de placa protectora de la zona ósea biopsiada, en el examen pre quirúrgico se percatan de un derrame pleural del lado izquierdo (líquido lechoso - quilotórax), fué drenado y requirió colocación de un tubo de drenaje pleural. Hospitalizada por 28 días y recibió octeótrido por 12 días, posteriormente remitida al HGP para completar tratamiento. Por episodios persistentes de quilotórax se hospitaliza por 2da vez en el HGP donde se realiza toracoscopía con clipado del conducto torácico.
Estado al ingreso: ingresa al servicio con TEP (triangulo de evaluación pediátrica) estable, lucida, orientada, colaboradora, afebril, eupneica.
Al examen físico: portaba VVC (vía venosa central), Aspecto respiratorio: crepitantes en base derecha, no sibilancias, porta tubo de drenaje pleural lado derecho, resto del examen físico sin particularidades. Se solicita analítica laboratorial (Hemograma, PCR, Perfil renal, Perfil hepático, electrolitos, Perfil lipídico, proteínas totales albumina, hemocultivo y urocultivo). En el perfil lipídico llamaba la atención colesterol total 202mg/dl, triglicéridos 252 mgdl, resto de los estudios dentro de rango, cultivos negativos. Se solicita además radiografías de control (Figura 1).
Evolución: Se retira tubo de drenaje pleural, se realiza interconsulta con sub especialistas, solicitan TAC y RMN de tórax, abdomen, pelvis y miembros inferiores, más densitometría ósea.
La TAC de tórax (Figura 2) informa atelectasia segmentaria a nivel de hilio basal, pequeño derrame pleural basal derecho. Llama la atención la disminución de la densidad ósea de la D12.
TAC de abdomen y pelvis disminución de la densidad más marcada a nivel de las vértebras lumbares con múltiples imágenes de aspectos osteolíticos de distribución difusa desde L1 a S2. (Figura 3) La afectación incluye a las palas iliacas y los alerones sacros, la cabeza femoral diáfisis proximal del fémur izquierdo, la afectación es tanto del hueso esponjoso como de la cortical Hacia el tercio medio del fémur se observa fisura patológica.
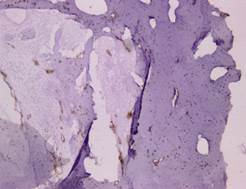
La densitometría ósea informa: valores densitométricos se encuentran en el límite inferior del rango normal esperado para la edad. Con estos datos, se plantea las posibles causas de osteólisis, desde el punto de vista renal: los resultados de gasometría, calcio y creatinina en orina espontanea fueron normales. Se realiza biopsia de fémur y aspiración medula ósea para inmunohistoquímica, la inmumarcación con S100 fue negativo y el D20-40 (Figura 4) y CD31, positivos.
Con estos resultados se descarta histiocitosis y se llega al diagnóstico probable de Síndrome de Gorham Stout.
En su 16 ddi, se inicia goteo de Pamidronato, el cual recibe por 3 días (inicia a 0,5mg/kp, luego 1mg/kp el 2do y 3er día, goteo para 4 horas), con buena evolución post goteo, fué de alta con planes de volver para nuevo goteo de Pamidronato en 3meses y recibir Sirolimus (alternativa terapéutica en pacientes pediátricos con anomalías vasculares y linfática)5.
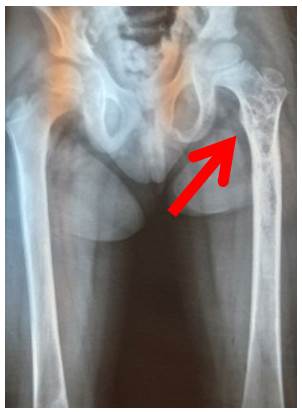
Figura 1 Radiografía de pelvis y miembros inferiores. La flecha muestra disminución de la densidad en región proximal de fémur izquierdo.

Figura 2 TAC de Tórax con contraste: La flecha muestra fina atelectasia segmentaria a nivel de hilio basal derecho.
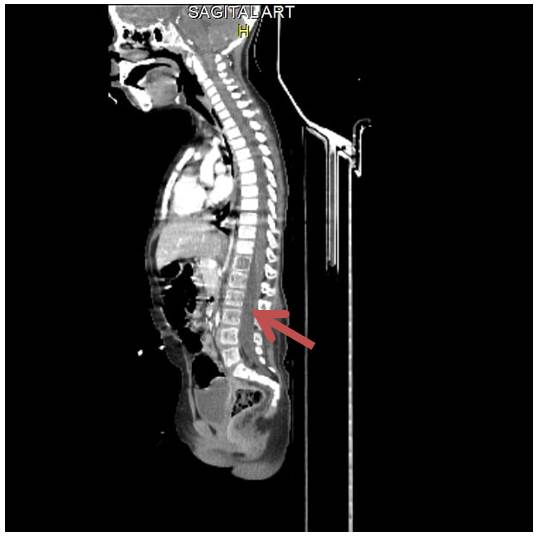
Figura 3 TAC de Vértebras: la flecha muestra disminución de la densidad, más marcada a nivel de las vértebras lumbares con múltiples imágenes de aspectos osteolíticos de distribución difusa desde L1 a S2.
DISCUSIÓN
La enfermedad planteó un desafío para los médicos a la hora de diagnosticar y tratar adecuadamente al paciente.
Presentamos el caso de una paciente de sexo femenino, de 5 años de edad, sin antecedentes familiares de interés.
El Síndrome de Gorham Stout no presenta asociación familiar o preferencia sexual y puede ocurrir en cualquier edad, pero es más común en adolescentes y adultos jóvenes6. La paciente del presente reporte, tenía un estudio previo de anatomía patológica de tumor de fémur sin hallazgos llamativos por lo que se descartó patología neoplásica, presentó además episodios recurrente de quilotórax espontáneo. El derrame pleural y el quilotórax pueden desarrollarse en el GSS, cuando la invasión linfoangiogénica se extiende a la cavidad pleural o al Conducto torácico1. El desenlace fatal suele estar relacionado con la presencia de quilotórax o inestabilidad espinal causada por destrucción osteolítica de las vértebras, con una mortalidad entre el 33 al 53%.
El GSS puede involucrar cualquier parte del esqueleto; sin embargo, el cráneo, el hombro y la pelvis son los sitios más comúnmente involucrados6. La paciente reportada presento afectación de la columna torácica, lumbar, pelvis y región proximal de fémur izquierdo, lesiones características compatibles con dicho síndrome.
Estudio de anatomía patológica con inmuhistoquímica que informaba material óseo focalmente positivos a la inmunomarcación con D2-40 y CD31, marcadores inmunohistoquímicos de células endoteliales linfáticas y proliferación de endotelio de vasos sanguíneos. Histopatológicamente, GSS se caracteriza por una arquitectura de proliferación anormal de linfáticos no neoplásicos, vasculares o tejido conectivo fibroso hiperplásico sin atipia celular, infiltración de células inflamatorias u osteoclastos7. Es decir la osteólisis masiva es acompañada de angiogénesis y linfangiomatosis, con proliferación de estructuras vasculares benignas y destrucción de matriz ósea2.
Diferencias en condiciones generales de salud, comorbilidades y el grado de pérdida de funciones implica diferentes tratamientos y enfoques4).
En relación al tratamiento, aunque no hay uno definido, incluye cirugía, radioterapia, e inhibidores de la actividad osteoclástica.
Concluimos que el caso clínico es compatible con el síndrome de Gorham Stout, la paciente recibió tratamiento con bifosfonato, (goteo de Pamidronato).