











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La fibrosis quística (FQ) es una patología de origen genético de herencia autosómica recesiva, causada por mutaciones en el gen que codifica la proteína reguladora de la conductancia transmembrana: Cystic fibrosis transmembrane conductance (CFTR, por sus siglas en inglés), que transporta activamente cloruro, bicarbonato y de manera secundaria agua. La condición causa alteraciones multisistémicas afectando principalmente al aparato respiratorio, el sistema digestivo, las glándulas sudoríparas y el conducto deferente1,2.
En Paraguay en el año 2003 se creó el Programa Nacional de Prevención de la Fibrosis Quística y del Retardo Mental. A partir del mismo se estableció la detección, diagnóstico y tratamiento de la FQ para todo recién nacido (RN) de forma obligatoria y gratuita. En el año 2016 se cambió la primera denominación por Programa Nacional de Detección Neonatal (PNDN)3,4,5.
Históricamente es importante recordar que a nivel mundial la inclusión de la FQ como una de las patologías a ser atendidas en los paneles de tamizaje neonatal ha sido un tema muy discutido, pues la misma no cumplía con requisitos esenciales para su inclusión, como por ejemplo contar con métodos de detección sensibles y tratamiento efectivo. Sin embargo con los avances en las técnicas de detección y los nuevos tratamientos esta patología fue incluida como parte de estos paneles. Estos factores influyeron en la decisión del PNDN de realizar, en sus inicios el tamizaje selectivo de FQ, a aquellos RN con síntomas o antecedentes familiares. Recién en el 2015 se logró dar cobertura a todas las muestras procedentes de las 18 Regiones Sanitarias (RS) del país4,5,6.
La incidencia de la FQ así como su detección en una población, depende de factores tales como el ambiente, el grado de consanguineidad de la población estudiada y de las variaciones fenotípicas derivadas del tipo de mutación (grupo étnico). Todas ellas responsables de las diferencias observadas en la población mundial, ejemplos de estas variaciones son las reportadas por Australia, país en donde la incidencia es de 1 por cada 2.500 RN mientras que en Japón se observa 1 caso por cada 100.000 a 350.000 RN. En Latinoamérica, Brasil presenta una incidencia de 1 en 1.600 en poblaciones con ascendencia europea y de 1 en 14.000 RN en afrodescendientes, mientras que en Argentina se observa una incidencia de 1 en 6.573 nacidos vivos. Por otro lado, en México la incidencia es 1 en 8.500 y en el Caribe, en Cuba la incidencia reportada es de 1 en 3.9007,8. La presencia de descendientes de origen africano es importante en Ecuador, Colombia, Venezuela y Brasil, aunque su porcentaje de forma global no supere el 10%. En Paraguay conforme a estudios genéticos la composición promedio del individuo paraguayo es la siguiente: 55,0 % europea; 38,0 % amerindia; 8,0 % africana4,7,9,8,10,11.
El objetivo del presente estudio fue conocer la prevalencia de FQ durante los años 2015, 2016 y 2017 en la población de recién nacidos de 0 a 28 días de vida, del Paraguay, cuyas muestras fueron remitidas al Programa Nacional de Detección Neonatal para su estudio.
METODOLOGÍA
Este es un trabajo descriptivo, retrospectivo de corte trasversal. Se analizaron los resultados de las muestras de RN enviadas al PNDN de enero del 2015 a diciembre del 2017, provenientes de los 1.063 sitios de toma de muestra (STM), distribuidos en las 18 RS.
La recolección de las muestras de sangre total se llevó a cabo por punción del talón de los RN en papel de filtro Whatman 903, secadas a temperatura ambiente por 2 a 4 horas y refrigeradas hasta su remisión al Programa, entre los 2 y 10 días posteriores a su extracción.
Criterios de inclusión: en relación a la edad, se consideró toda muestra de RN hasta 28 días de vida y la calidad consistió en que la gota de sangre tenga un mínimo de 0,5 cm de diámetro, de distribución homogénea a ambos lados del papel.
La técnica utilizada en laboratorio consistió en la cuantificación de la tripsinainmunoreactiva (TIR), por el método de fluoroinmunoensayo en tiempo resuelto (DELFIA/PERKIN ELMER’S) en muestras de sangre seca recogidas en papel de filtro. El valor de referencia hasta 55mgr/dl.
Para la determinación de la positividad en el PNDN se utiliza la estrategia de trabajo TIR(+)/TIR(+), que consiste en la realización de una primera prueba de la tripsinainmunoreactiva (TIR) y la solicitud de una segunda muestra a todos los casos que resultarán positivos en la primera. En caso de que ambas muestras presentaran resultados alterados, la confirmación diagnóstica se llevo a cabo mediante el test del sudor, que consiste en la medición del cloro por el método de titulación coulométrico en muestras de sudor. A los RN con edades superiores a los 30 días de vida al momento de la segunda muestra se les realizó de forma directa el test del sudor12.
Para el análisis de la base de datos se utilizó estadística descriptiva en una planilla Excel.
RESULTADOS
De enero del 2015 a diciembre del 2017 se recibieron 263.312 nuestras de recién nacidos, provenientes de los 1.063 sitios de toma de muestra distribuidos en las 18 RS, la distribución por año de acuerdo a las muestras colectadas, las procesadas, los porcentajes de las mismas y los casos confirmados se observan en la Tabla 1.
Tabla 1 Número de muestras recibidas/estudiadas y casos de FQ detectados por año.
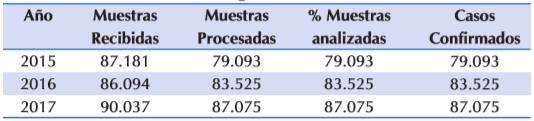
Fuente: Datos del PNDN (no publicados)
Del este total de muestras analizadas en año 2015, 80 cumplieron la estrategia TIR/TIR, de las cuales en el 15% (12/80) se confirmo FQ. Para el 2016, de 98 cumplieron la estrategia el 20,4%(20/98) fueron confirmadas y finalmente en la año 2017 de los 120 que cumplieron la estrategia el 14%(17/120) fueron confirmados. Es importante recalcar que 13 de los RN que tuvieron alterada la TIR en la primera determinación fallecieron Tabla 2
Tabla 2 Distribución de casos confirmados de FQ e incidencia por año.

# RN fallecidos antes de la confirmacion diagnóstica.
TEST del SUDOR: Pacientes IRT/IRT fuera de rango+Pacientes con más de 30 dias de vida+Pacientes con sospecha de FQ por el test del piecito.
En la Tabla 3, se presenta el resumen del Nº de nacimientos registrados por año, así como la sumatoria de estos. El número de STM, las muestras recibidas y analizadas en el periodo de tiempo del presente estudio y los casos confirmados de FQ, por RS.
Tabla 3 Número de Nacimientos registrados-muestras recibidas/estudiadas y casos de FQ detectados por Región Sanitaria del 2015 al 2017.
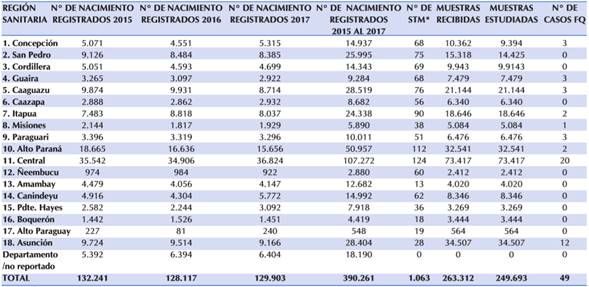
Fuente: http://www.dgeec.gov.py/ - Datos del PNDN (no publicados)
Así también del total de nacimientos registrados a nivel país, durante estos tres años el porcentaje de cobertura para el estudio de la FQ aumento de un 60 a un 67% (Figura 1).
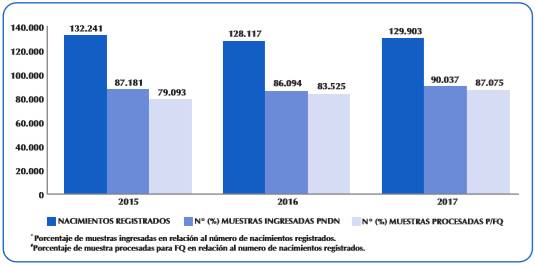
DISCUSIÓN
La detección neonatal de la FQ en Paraguay ha sido garantizada por Ley desde el año 2003, la cobertura ha ido aumentando de forma paulatina, pudiéndose desde el 2015 contarse con los recursos suficientes no solo para absorber los costos del tratamiento de los pacientes diagnosticados con anterioridad y de los nuevos, sino para tamizar al 100% de las muestras recibidas, permitiéndonos el análisis de la incidencia de la patología en la población paraguaya de RN. En este trabajo se reporta por primera vez las incidencias anuales de la FQ en el Paraguay por tres años consecutivos, siendo estas similares a las descriptas en otros países sudamericanos, que oscilan entre 1/3000 a 1/ 85008,11.
La importancia de conocer la incidencia de FQ en nuestro país, no solo permite la proyección y el abastecimiento para la provisión en tiempo y forma de los medicamentos e insumos a ser entregados a los afectados, sino que además refuerza la importancia del diagnóstico precoz ante la evidencia demostrada en los resultados clínicos a corto y largo plazo en personas diagnosticadas por tamizaje neonatal13.
La mayor diferencia entre las muestras ingresadas y procesadas se observó durante el primer año de universalización de la pesquisa para FQ, diferencia que disminuyó considerablemente los años siguientes, para lo cual se llevaron a cabo capacitaciones y envío de materiales informativos a los encargados de los STM.
La discrepancia observada entre el número de recién nacidos y el número de muestras recibidas correspondientes a la 18 RS (Asunción), podría deberse a diversos motivos como los partos domiciliaros estimados en poco más de 2.500 según el último reporte publicado por la Dirección General de Estadísticas Encuestas y Censos (DGEEC), y nacimientos que se produjeron en otra RS pero cuyas muestras fueron extraídas en STM de la capital, pasando a engrosar el número de muestras recibidas y estudiadas14.
Este estudio tiene la limitación de no conocerse las mutaciones genéticas de los pacientes, teniendo en cuenta los casos de atípicos de FQ que corresponden a mutaciones del CFTR que se presentan con pocos o ningún síntoma durante la niñez e incluso en la edad adulta 13,15. Sin embargo, al ser el primer estudio epidemiológico sobre la incidencia de FQ en el país, aporta datos que hasta la fecha no se conocían sobre esta afección.

