











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La Arteritis de Takayasu (AT), o "enfermedad sin pulso", o Síndrome de Martorell, es una vasculitis crónica de los grandes vasos, de origen desconocido, que afecta predominantemente a la Aorta y sus ramas principales. La edad de inicio en pediatría más frecuente corresponde a la franja entre 10 y 18 años, con predominio del sexo femenino1,2.
En el 2008, el grupo de trabajo de vasculitis de la Sociedad Europea de Reumatología Pediátrica (PRES), la Liga Europea Contra el Reumatismo (EULAR), y la Organización Internacional de Ensayos de Pediatría Reumatología (PRINTO), establecieron los criterios de clasificación para las vasculitis en la infancia, mencionando entre las más frecuentes a la Púrpura de Henoch-Schonlein, la Enfermedad de Kawasaki, la Poliarteritis Nodosa infantil, la Granulomatosis con Poliangeitis infantil (Granulomatosis de Wegener), la AT infantil, entre otras (Tablas 1 y 2)3,4,5.


La AT es rara y difícil de diagnosticar. Los diagnósticos diferenciales de las vasculitis sistémicas y entre ellas la AT, incluyen lo descripto en la tabla 3 6.
Para evaluar la actividad de la enfermedad en pacientes con AT se utilizan los estudios establecidos por Kerr et al (National Institutes of Health). Nueva presentación o empeoramiento de 2 o más de los siguientes datos indican enfermedad activa7:
Síntomas sistémicos, como fiebre y artralgias (sin causa identificada)
Velocidad sedimentación globular acelerada
Características de isquemia o inflamación vascular, como claudicación, pulso disminuido o ausente, soplo, carotodinia o presión arterial asimétrica en los miembros superiores o inferiores (o ambos)
Características angiográficas típicas
Para el diagnóstico y seguimiento de la enfermedad, son necesarios estudios de imágenes como Ecografía con doppler, angiografía convencional, angio-RMN, angio-TAC, pudiendo sumarse PET con fluorodesoxiglucosa. La aorta torácica y abdominal con los grandes vasos son los más afectados en AT juvenil 1,4,8,9,10,11.
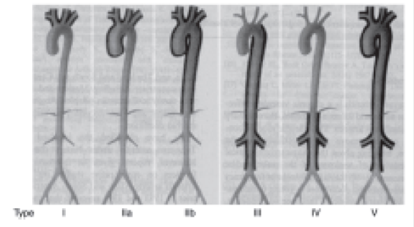
La AT se divide en cuanto a los hallazgos angiográficos y se definieron por la Clasificación angiográfica de Hata (Tokio, 1994) (Figura 1)11.
La AT es una vasculitis poco frecuente en niños, por lo que presentamos el caso de un paciente con AT avanzada.
CASO CLÍNICO
Paciente masculino de 13 años de edad, con antecedente de astenia de 1 año de evolución, síncopes en varias ocasiones de 4 meses de evolución, pérdida de peso, claudicación intermitente y dificultad respiratoria progresiva, que se reagudizan en las últimas horas. Acude en varias ocasiones con facultativos, recibiendo tratamiento sintomático. Ingresa hipoactivo, hipotrófico, con palidez marcada y sudoración profusa.
Examen físico: extremidades superiores frías y sin pulsos palpables; miembros inferiores tibios con pulsos palpables, en donde se constata cifras tensionales elevadas (240/70mmHg). Área cardíaca: Ictus cordis se ve y se palpa, presenta frémito en foco aórtico y carótida derecha, soplo sistólico en foco aórtico grado IV/VI que irradia a ambas carótidas.
Laboratorio: Glóbulos Blancos 8.200, Neutrófilos 68%, Linfocitos 32%, Hemoglobina 9,9mg/dl, Hematocrito 32,3%, Plaquetas 345.000, PCR + 24 (<6), VSG 70mm, urea 23, creatinina 0,61, GOT 14, GPT 10, TROPONINA I <0,01, Orina simple normal. C3 197,8mg/dl (90-180), C4 27,6mg/dl (10-40), ANA negativo, ANCA-MPO negativo, ANCA-PR3 negativo, anticuerpos para Sx Antifosfolipidico negativos.
Ecocardiografía-doppler: hipertrofia concéntrica del ventrículo izquierdo, insuficiencia aórtica leve a moderada, disminución del calibre de aorta transversa y descendente de 9mm y a nivel de aorta abdominal se observa rama que emerge midiendo 3mm con gradiente superior a 100mmHg.
Eco-doppler renal: estenosis hemodinámicamente significativa en el tercio medio de la arteria renal derecha y ostium de la arteria renal izquierda.
Angio-TAC: estenosis de aorta, ramas abdominales y carotídeas (Figura 2: a-b-c).
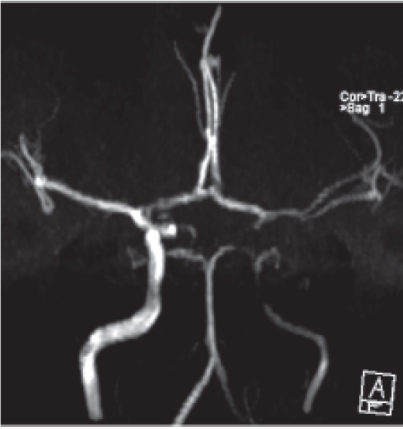
Angio-RMN cerebral: afectación de todo el trayecto intracraneano de arteria carótida interna izquierda y reducción del calibre de ambas arterias cerebrales (Figura 3).
Se confirma el diagnóstico de Arteritis de Takayasu, tipo V. Tratamiento: bolos de metilprednisolona continuando con prednisona, goteo de ciclofosfamida EV, además de goteo de inotrópicos (dobutamina) por 72 horas por choque cardiogénico con mejoría y posteriormente amlodipina + carvedilol. Ante evolución clínica favorable, es dado de alta tras dos semanas de internación, con seguimiento ambulatorio.
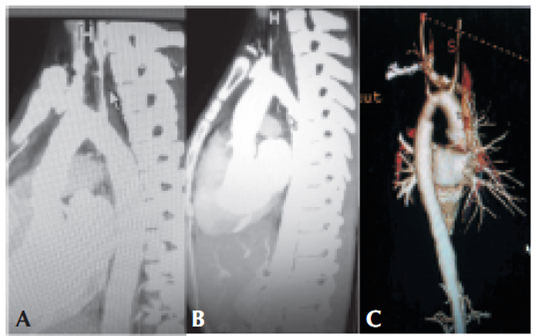
Figura 2 Angio-TAC de grandes vasos: A: estenosis de subclavia izquierda (marcada con flecha). B y C: estenosis de aorta.
DISCUSIÓN
La Arteritis de Takayasu es una vasculitis de grandes vasos, que se caracteriza por inflamación granulomatosa crónica de la pared del vaso, de etiología idiopática, con múltiples implicancias genéticas, con estudios que relacionan al HLA B*52, DRB1*1502, DRB5*0102, DQA1*0103, DQB1*0601, DPA1*02-DPB1*090, en pacientes de origen oriental, como pronóstico de mayor gravedad12,13. Vargas-Alarcón et al demostraron HLA B*15, B*39 y B*40 en la mayoría de sus pacientes mexicanos14. Sin embargo, Salazar et al describen presencia de HLA-DRB1*1602 and HLA-DRB1*1001 en una población colombiana15. No pudimos realizar estos estudios en nuestro paciente en el país.
Watts et al encontraron en el registro del Reino Unido: 6 pacientes (5 mujeres), <40 años en la presentación en el 43% entre 2000 y 2005, con una incidencia anual en <40 años de 0.3/millón16. Soto et al publicaron pacientes mestizos mexicanos: un total de 94 pacientes (85%) fueron mujeres y 16 (15%) hombres, con una relación hombre: mujer de 1:5.8. La edad media al diagnóstico fue de 26±9 años. Al momento del diagnóstico, 101 pacientes (91%) eran <40 años de edad17. Clemente et al en un estudio en Brasil encontraron: 71 pacientes con AT juvenil, donde 51 (71,8%) eran niñas. Las medias de edad de inicio de los síntomas y del tiempo hasta el diagnóstico fueron de 9,2 (±4,2) años y 1,2 (±1,4) años, respectivamente18. Kerr et al. incluyeron 30% de pacientes pediátricos en su estudio y reportaron una incidencia en todas las edades de 2.6/1 000 0007. No contamos con registros a nivel nacional.
Al inicio de la enfermedad, pueden aparecer síntomas constitucionales inespecíficos como fiebre, malestar general y pérdida de peso. Posteriormente, la inflamación de las arterias involucradas progresa, con estenosis segmentaria, oclusión, dilatación y/o aneurisma. Esto puede causar dolor en las extremidades, claudicación, soplos, ausencia o disminución de pulsos y pérdida de la presión arterial. Afecta predominantemente al arco aórtico y sus ramas primarias, aorta ascendente, aorta descendente torácica y aorta abdominal. Generalmente sigue un curso insidioso, sin embargo, también puede ocurrir presentación con pérdida visual aguda o accidente cerebrovascular. En AT juvenil, las manifestaciones neurológicas incluyen ACV (17%) y cefalea (31%). Las manifestaciones dermatológicas son raras, y se describen rahs y nódulos subcutáneos. La presentación más frecuente en pediatría es la hipertensión arterial (82.6%), continuando con cefaleas (31%), fiebre (29%), disnea (23%), pérdida de peso (22%), vómitos y dolor abdominal (20.1%)12,18,19,20,21. La afección osteoarticular es rara en niños, describiéndose solo en 14% de niños con AT, aunque en Sudamérica se presenta hasta en 65% de los casos12. Nuestro paciente no refirió dolores articulares.
No existen marcadores de laboratorio específicos para esta patología. En cuanto a reactantes de fase aguda: el 53% de los niños tienen una VSG acelerada, comparada con el 45% de los adultos con AT, considerándose a la VSG como el mejor indicador de actividad en adolescentes, aunque puede seguir elevado en la remisión de la enfermedad. La PCR aparenta relacionarse bien con la actividad de la enfermedad22,23,24,25. Se describen otros marcadores como: activador tisular del plasminógeno, molécula de adhesión intercelular 1, molécula de adhesión vascular 1, selectina E, molécula de adhesión plaquetaria endotelial 1, anticuerpos anti-célula endotelial, anticuerpos anti-monocito, anticuerpos dirigidos contra anexina V26,27,28,29,30.
En el trabajo de Soto et al17, la enfermedad aórtica generalizada (Tipo V) se encontró en 76 (69%) pacientes, la enfermedad tipo I estuvo presente en 21 (19%), tipo IIa en 3 (3%) mientras que tipo IIb en 4 (4%); Tipo III también se encontró en 4 casos (4%) y Tipo IV en 2 (2%). Nuestro paciente presenta una clasificación Tipo V, coincidente con la presentación más frecuente.
El tratamiento de la AT se centra en controlar la inflamación y manejar las complicaciones, por lo que depende de la presentación clínica. Los corticoides son el grupo de medicamentos más utilizados para controlar la enfermedad activa y lograr la remisión, pudiendo usarse bolos de Metilprednisolona y corticoides orales a dosis altas para luego continuar con descenso progresivo, por largo tiempo. Se utilizan además otros inmunosupresores ahorradores de corticoides como agentes citotóxicos (ciclofosfamida, metotrexato, azatioprina, micofenolato)7,31,32,33,34, y en casos de poca respuesta a los mismos: agentes biológicos como tocilizumab (anti-IL 6)35, Rituximab (anti CD20 expresado en la superficie de linfocitos B)36, anti-Factor de Necrosis Tumoral (FNT) como etanercept, infliximab, adalimumab34,37,38. Los procedimientos cardiovasculares descriptos con más frecuencia son cirugía de injerto (bypass), angioplastia percutánea con balón, angioplastia y stents, recorte de aneurisma y revascularización, con diferentes resultados7,39,40,41.
Como puede observar, la AT es una vasculitis potencialmente mortal, y debe sospecharse en un niño que presenta síntomas de hipertensión, fiebre, pérdida de peso, vómitos, cefalea. La presunción obliga a la realización de imágenes vasculares, a fin de llegar al diagnóstico temprano y al tratamiento oportuno.

