










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPediatría (Asunción)
On-line version ISSN 1683-9803
Pediatr. (Asunción) vol.43 no.2 Asunción Aug. 2016
https://doi.org/10.18004/ped.2016.agosto.137-144
CASO CLÍNICO
Encefalitis de Bickerstaff. Reporte de caso y revisión de la literatura
Bickerstaff’s encephalitis. Case report and literature review
Joel Eladio González López (1), Laura Rojas de Recalde(1), Luis Chamorro Noceda(2)
1 Cátedra y Servicio de Neurología. Universidad Católica Nuestra Señora de la Asunción, Facultad de Ciencias de la Salud. Hospital Central, Instituto de Previsión Social. Asunción, Paraguay. Médico Residente III año de Neurología y Médica Pediatra-Neuróloga infantil.
2 Cátedra y Servicio de Pediatría. Universidad Católica Nuestra Señora de la Asunción, Facultad de Ciencias de la Salud. Hospital Central, Instituto de Previsión Social. Asunción, Paraguay. Médico Pediatra, Jefe de sala.
Correspondencia: Dr. Joel E. González López. E-mail: joelgon0010@hotmail.com
Conflicto de intereses: Los autores declaran no poseer conflicto de intereses.
Recibido: 05/03/2016; Aceptado: 20/07/2016.
RESUMEN
Se presenta un caso de encefalitis de Bickerstaff (EB), en un niño de 4 años de edad con antecedente de dos internaciones en el Servicio de Pediatría del Hospital Central del Instituto de Previsión Social (HC.IPS): en la primera ocasión, por síndrome febril prolongado y la segunda, dos semanas después, por un síndrome neurológico con cuadro clínico de somnolencia, mareos y como síntomas iniciales: oftalmoplejía y ataxia; a los que se agregaron el compromiso de otros pares craneales. Le antecedieron una sinusitis etmoidal e impétigo en el primer ingreso. Lo llamativo del caso es el resultado positivo para Salmonella Thyfi en la evaluación serológica realizada ante el síndrome febril prolongado del citado paciente. La evolución fue satisfactoria con recuperación funcional total a los 60 días. Se hace una revisión del caso y de los síndromes asociados a anticuerpos antigangliósidos en especial a los anti GQ1b.
Palabras clave: Encefalitis de Bickerstaff, síndrome de Miller Fisher, anticuerpo anti GQ1b.
ABSTRACT
A Bickerstaff`s case brainstem encephalitis (EB).It was in a child of 4 years of age. He had two hospitalizations in the pediatrics service of Hospital Central Institute of Social Security (HC.IPS): the first by prolonged fever and second, two weeks later by a neurological syndrome characterized by drowsiness and dizziness as initial symptoms, ophthalmoplegia and ataxia; those who were adding the involvement of other cranial nerves. Respiratory symptoms (ethmoid sinusitis) and skin lesions (impetigo) were the infectious antecedents identified in the first entry, to which test positive for salmonella thyfi was added in serological evaluation by a history of prolonged febrile syndrome. The evolution was satisfactory with full functional recovery at 60 days. A revisions of the case and of the syndromes associated with antiganglioside antibodies especially the anti GQ1b was made.
Keywords: Bickerstaff’s encephalitis, Miller Fisher syndrome, anti GQ1b.
INTRODUCCIÓN
En el año 1951 Bickerstaff y Cloake informaron tres casos de una entidad nosológica caracterizado por oftalmoplejía, ataxia y bajo nivel de conciencia y plantearon que la lesión subyacente se hallaba en el tallo cerebral(1). El cuadro con estas características fue denominado como encefalitis del tallo cerebral de Bickerstaff. Es una entidad neurológica, grave en su inicio, post infecciosa, inmunomediado, generalmente monofásica y con un pronóstico usualmente benigno, con recuperación espontánea en la mayoría de los casos(2-4). Los criterios clínicos en cierto modo estrictos son la oftalmoplejía externa, la ataxia y la alteración de la conciencia o hiperreflxia(1,3,5,6).
En la década de los noventa investigadores japoneses usando métodos de inmunohistoquímica describieron tanto en el síndrome de Miller Fisher(SMF) como en la (EB) una asociación con anticuerpos antiganglíosidos GQ1b y postularon que ambas enfermedades eran variantes de una misma entidad en razón al perfil clínico e inmunológico que los caracterizan y los denominaron en consecuencia ¨Síndrome anti GQ1b¨ (1-5,7-10).
La lesión localizada en el sistema nervioso central puede presentarse aislada, o superpuesta a un compromiso del sistema nervioso periférico, específicamente al SMF en cuyo caso se habla de síndrome Fisher - Bickerstaff(1-5).
Algunos autores postulan que de existir compromiso del nivel de conciencia es más adecuado la denominación directa de EB(5).
Existen muy pocos reportes de la EB en niños y la mayoría de las descripciones están documentadas sobre casos en adultos.
PRESENTACIÓN DEL CASO
Niño de 4 años, peso 19,700 grs. y talla 115cm, procedente del área metropolitana (Fernando de la Mora). Presenta ingresos en el Servicio de Pediatría del Hospital Central del Instituto de previsión Social (HC-IPS), en el mes de agosto y setiembre del 2015 respectivamente.
La primera ocasión, en la sala de infectología por síndrome febril prolongado fue del 14 al 24 de agosto del 2015, y sale de alta con los diagnósticos de: Sinusitis etmoidal, Impétigo, y anemia leve normocítica, normocrómica. El laboratorio para el despistaje bacteriológico del síndrome febril prolongado dio como resultado una serología positiva para Salmonella Typhi, sin cuadro gastroenterológico clínico. El cuadro febril volvió a presentarse en su domicilio oscilando entre 37,8 y 38 grados en forma irregular sin otros signos durante seis días y luego cedió de manera espontánea, motivo por el que la madre ya no volvió a la consulta
El segundo ingreso, es en la sala de polivalente, 12 días después del alta anterior. El cuadro clínico se inicia 5 días antes del ingreso con somnolencia marcada y mareos como síntomas iniciales, marcha atáxica (andar inestable como de ebrio), ojos desviados hacia adentro, más evidente en el lado derecho y dificultad para articular varias palabras de modo gradual. Sin fiebre ni otros signos/síntomas.
Al internarse, la evaluación del sensorio tiene en una escala de coma de Glasgow, de 14-15/15 alternante ( un despertar intercalado con somnolencia prolongada), debilidad muscular leve en los 4 miembros, con mayor compromiso en los inferiores, ataxia de tronco y durante la marcha, reflejos osteotendinosos normales, oftalmoplejía bilateral asimétrica, que predomina en el lado derecho. También presentaba disartria.
Los diagnósticos al ingreso por la guardia fueron: síndrome cerebeloso de etiología a determinar. Sospecha de Síndrome de Miller Fisher.
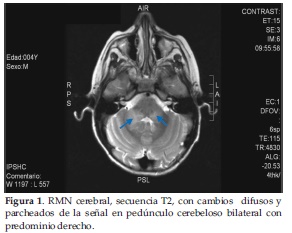
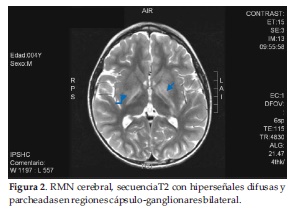
Se le realizó RNM cerebral simple del 05/09/15 (quinto día de enfermedad y primer día de ingreso) que fue informado por el radiólogo de la siguiente forma: lesiones difusas y parcheadas que afectan a la región ganglio basal, tronco encefálico, pedúnculos cerebelosos medios y médula espinal, sugestivo de encéfalo mielitis en primer lugar, desmielinizante? Inflamatorio-infeccioso?, con sugerencias a corroborar con la semiología evolutiva y tras la administración de contraste (Figura 1 y 2).
La Punción Lumbar 6/09/15; (sexto día de enfermedad), mostró el siguiente resultado:
Aspecto: límpido
Color: Incoloro
Color del sobrenadante: incoloro sin botón hemático
Coagulación: ausente
Leucocitos: no se observan
Hematíes: no se observan
Glucosa: 70 mg/dl
Proteínas: 13mg/dl
Todos los otros estudios: hemograma, química sanguínea, perfil colagénico, dosaje de hormona tiroidea, uroanálisis y otros fueron normales.
En estas condiciones, 3 días después y con evolución estable, es trasladado a la sala de medicina Interna, unidad de neuropediatría.
En esta sala (8/09) el reinterrogatorio a los padres detalla una historia similar al del ingreso.
Al examen se observó un nivel de conciencia fluctuante, SCG 14-15/15, con periodos de somnolencia cada vez más cortas y vigilia bien activa.
Llamó la atención la ligera debilidad de los miembros, con predominio en los inferiores. La fuerza muscular evaluada por la escala del Medical Research Council (MRC) fue de 3-4/5 en los segmentos distales; 4/5 en los segmentos proximales de los miembros inferiores, 4/5 en los segmentos proximales y distales de los miembros superiores. Sin dismetría ni adiadococinesia. Ataxia del tronco y de la marcha, oftalmoplejía externa bilateral asimétrica con mayor compromiso en el lado derecho, disartria y se agregaron diplejía facial y trastorno para la deglución en esta sala. Los reflejos osteotendinosos: normales (++/++++), Babinski ausente, Romberg presente. Sin signos meníngeos ni de hipertensión endocraneana. Sin compromiso de la musculatura respiratoria, nivel sensitivo, ni compromiso esfinteriano.
En base a la historia y al examen caracterizado por la alteración del nivel de conciencia y mareos desde el inicio, oftalmoplejía externa relativamente simétrica y ataxia, con compromiso de otros pares craneales del tallo cerebral y la RNM cerebral patológica se planteó el diagnóstico de una encefalitis del tallo cerebral probablemente autoinmune atendiendo que estuvo hospitalizado semanas antes por un cuadro infeccioso.
Los estudios de anticuerpos anti-gangliósidos no se realizaron porque el Servicio no dispone de laboratorio para esa línea de investigación.
La Electromiografía y Velocidad de conducción (11/09/15), 11 días de enfermedad, concluye ligera disminución de las velocidades de conducción motriz. La onda F se halla a 28 ms de latencia. Las latencias distales se hallan dentro del rango. El registro muestra actividad de tipo neurogénico con unidades polifásicas y desgarradas. Diagnóstico: Polineuropatía desmielinizante.
El cuadro clínico en la objetivación diaria desde el inicio del evento ha demostrado una evolución progresiva en severidad en cuanto a la ataxia y los trastornos deglutorios fundamentalmente, pero no agresiva. No obstante se administró inmunoglobulina intravenosa 1gr/Kg/día por 2 dias con buena tolerancia.
El paciente se mantuvo estable, no hizo complicaciones.
Sale de alta el 14/09/15, completando una internación de 9 días, con mejorías en la estabilidad del tronco y la marcha y los otros signos estables, es decir sin cambios.
El Diagnóstico al alta fue el de Encefalitis del tallo cerebral de Bickerstaff.
Se argumentó este diagnóstico en base a la clínica que debuta con alteración del nivel de conciencia (que no forma parte del síndrome de Miller Fisher), y que evolucionó a una oftalmoplejía y ataxia más que a una debilidad muscular propiamente dicha y compromisos de otros pares craneanos del tallo cerebral. La RNM con signos inflamatorios del tallo cerebral y el LCR sin disociación albumino-citológica. Todo esto precedido por un cuadro infeccioso y con una evolución favorable.
El mejoramiento del paciente, en la sala, se observó primeramente en la recuperación del nivel de conciencia, la ataxia del tronco y de los miembros y la oftalmoplejía, precisamente en los signos que marcaron el inicio de la enfermedad.
Los signos que persistieron hasta la cuarta semana de evolución (consulta de control), aunque con mínima expresión fueron la disartria para algunos fonemas, la diplejía facial para esfuerzos extremos y la dificultad para la deglución de líquidos generándose eventos de tos, no así con los alimentos sólidos. La recuperación funcional total se observó a los 60 días.
DISCUSIÓN
La encefalitis de Bickerstaff se caracteriza por la triada de oftalmoplejía bilateral relativamente simétrica, ataxia y alteración del nivel de conciencia o hiperreflexia(1,3,4,7,11-13), y el síndrome de Miller Fisher por la triada de oftalmoplejía, ataxia y arreflexia o hiporreflexia (3-5,7,9,10,14-17).
Algunos autores refieren que los pacientes que presentan hiperreflexia no tienen alteración de la conciencia (13,17) por eso en la definición se presenta las dos alternativas: alteración de la conciencia o hiperreflexia. Otros autores sin embargo mencionan que ambas condiciones pueden estar presentes en un mismo paciente (9).
El caso que se presenta coincide con la mayoría de los casos publicados en la literatura médica. Predominó la alteración de la conciencia, sin hiperreflexia y los reflejos osteotendinosos estuvieron normales.
En 1951, Bickerstaff y Cloake informaron de una entidad caracterizada por alteración de la conciencia, oftalmoplejía y ataxia, planteando que la lesión se hallaba en el tallo cerebral y en 1956 Miller Fisher describió tres casos de oftalmoplejía, ataxia y arreflexia con lesión en el nervio periférico y señaló una relación con el síndrome de Guillain-Barré por la disociación albumina citológica observada. En la década de los noventa al describirse la presencia de anticuerpos anti GQ1b en el 68% de la EB y en el 83% del SMF se postuló que ambas entidades eran variantes de una misma enfermedad autoinmune (1,3,4,9,12,15). En este punto en el año 2001 Odaka y col. introdujeron el término de ¨Síndrome GQ1b¨ para referirse a diferentes morbilidades donde puede encontrarse el anticuerpo IgG anti GQ1b sugiriendo un mecanismo antigénico común (4,6,18,19).
Se cita en este grupo al SMF, la oftalmoparesia aguda (OA), la EB y el síndrome de Guillain Barré como parte del espectro continuo de este síndrome (3,4,6,9,12,15,20).
El Laboratorio del Servicio de la Seguridad Social no tiene aún incorporado las investigaciones en la línea del dosaje de los niveles de anticuerpos antigangliósidos, razón por lo que el paciente no fue sometido a este estudio.
Se estima que la incidencia anual es de 0.078 casos por cada 100.000 habitantes con un ligero predominio en el sexo masculino. Relación hombre: mujer1.3:1.Con este dato su prevalencia e incidencia es menor que la del Síndrome de Guillain Barré y SMF (1-5,7,11,15,20).
En el país no hemos encontrado una publicación anterior a la presentada, en la población infantil.
Se describe un cuadro infeccioso precediendo al evento neurológico como inductor de la formación de anticuerpos IgG anti GQ1b(5,14,15,21). Algunos autores describen hasta un 66% a las infecciones de vías respiratorias como antecedente del evento neurológico(3).
En el caso que describimos el paciente estuvo internado por una patología infecciosa febril prolongada, unas semanas antes del cuadro neurológico, que resultó ser una afección aguda y mixta, aparentemente superpuesta a un síndrome febril prolongado. Una, de base respiratoria (la sinusitis etmoidal), la otra, una afección cutánea (Impétigo); no hubo aislamiento de gérmenes en ambos cuadros. La investigación serológica para el síndrome febril prolongado resultó positiva para salmonella typhi sin signos gastrointestinales en ningún momento de la historia, previa a esta primera internación.
En la literatura médica consultada, se menciona al Campylobacter yeyuni y al Haemophilus influenza como infecciones más frecuentemente asociadas comparado con grupos controles como agentes con alta capacidad inductora de reacción antígeno anticuerpo anti GQ1b en pacientes sensibles(1,4-6,11,13). Pero también se han reportado casos de EB con antecedente de infecciones por herpes simple, citomegalovirus, virus del Epstein Barr, virus de la varicela zoster, virus del sarampión, salmonella typhi, parathyfi A,B o C, mycoplasma pneumonie y staphylococcus aureus(1,2,5,8,11,13,16,22-24).
En el caso reportado, algunos de los agentes causales de cualquiera de los 2 cuadros agudos febriles así como la salmonella typhi asintomática podrían estar implicados, por formar parte del listado anteriormente citado.
Bickerstaff postuló un efecto secundario más que un efecto directo del agente bacteriano como causa probable de la presentación neurológica(5).
La patogénesis estaría explicada por un mimetismo molecular mediado por auto anticuerpos anti GQ1b debido a que existen oligopolisacáridos en ciertos micro organismos que al inducirse la producción de anticuerpos contra estos agentes desarrollan una reacción cruzada en pacientes sensibles y que en ciertos tejidos nerviosos diana se expresan de manera relevante el antígeno GQ1b alterando la función de estas estructuras(5). Este hecho sugiere una base fisiopatológica autoinmune de la enfermedad(1-6,9,10,12,13). Las áreas que expresan en forma relevante el antígeno GQ1b son los paranodos (región citoplásmica de mielina que es adyacente al nodo de Ranvier) de los nervios oculomotores, husos musculares de las extremidades, en los ganglios de las raíces dorsales y el sistema reticular ascendente del tallo cerebral(4,6,7,20). Del mismo modo se expresa en forma importante en los nervios glosofaríngeos y del vago lo que explica la parálisis orofaríngea en este síndrome(6), así como en el nervio facial, en el ganglio ciliar o la placa terminal en el músculo esfínter-pupilar produciendo midriasis(6).
El anticuerpo anti GQ1b se liga también a la unión neuromuscular, causando una masiva liberación de acetilcolina desde las terminales nerviosas produciendo bloqueos en la conducción(4,6,8).
En cuanto al compromiso de los pares craneales adicional a la oftalmoplejía externa, nuestro paciente presentó parálisis facial bilateral, disartria y trastornos deglutorios. Esto demuestra que gran parte de los nervios craneales que expresan en forma relevante el antígeno GQ1b estuvieron afectados. Cabe destacar también que en nuestro paciente el trastorno deglutorio fue uno de los signos con mayor presencia temporal e intensidad de afectación. Fue el último trastorno que desapareció, luego de 60 días de permanencia.
De hecho en la práctica clínica y en las descripciones originales se mencionan los otros signos y síntomas que pueden presentarse tanto en el síndrome de Miller Fisher como en el de Bickerstaff tales como la ptosis, midriasis, parálisis facial, trastorno en la deglución, disturbio sensitivo periférico y otros signos bulbares(1,2,5,6,11,13); en el caso de la EB pueden presentarse además signos de afectación de las vías largas como la respuesta plantar extensora o signo de Babinski y/o pérdida hemisensitiva(7,13,14,25). Todo esto asociado a la pérdida de conciencia, ataxia y oftalmoplejía.
En la encefalitis de Bickerstaff la alteración de la conciencia exhibe grados variables de presentación, (somnolencia, sopor, estupor y coma) sugiriendo un compromiso del sistema activador reticular del tallo cerebral, probablemente secundario a la permeabilidad defectuosa de la barrera hemato-encefalica en ciertas regiones como el área postrema, permitiendo el pasaje de los anticuerpos y su acción sobre los antígenos GQ1b del tallo(4,5,12). En nuestro caso el niño debutó con hipersomnolencia y mareos cinco días antes que la ataxia y la oftalmoplejía se instalaran.
En el paciente que presentamos la debilidad muscular no fue muy relevante como fue la ataxia. Su calificación en cuanto a la fuerza muscular en el segmento más afectado fue de 3-4/5. En relación al trastorno para la marcha, si el paciente manifiesta remarcable debilidad (parálisis) con un valor de tres o menos para la escala de Fuerza Muscular del MRC el caso debe ser considerado como una encefalitis de Bickerstaff asociado o superpuesto al síndrome Miller Fisher o Guillain Barré(6). Los reflejos osteotendinosos en todos los puntos se mantuvieron en ++/++++, es decir normal. Esto coincide también con la mayoría de las publicaciones sobre el tema y en la presencia generalmente de uno de los dos signos dentro de la triada: alteración de la conciencia o hiperreflexia(1,3,4,7,11-13); en el caso nuestro fue alteración de la conciencia.
El diagnóstico se realiza fundamentalmente ante una alta sospecha por los signos clínicos típicos y la exclusión de otros diagnósticos, a través de estudios complementarios en los Servicios como el nuestro donde no se realizan estudios de anticuerpos antigangliósidos(1,5,6,20).
Respecto a la neuroimagen, la RNM cerebral es el estudio ideal porque es mucho más sensible para identificar signos inflamatorios o edema del SNC. El momento útil para su realización es entre el primer y tercer día de inicio de la enfermedad(2,13). Se describen lesiones difusas o parcheadas hipointensas en T1, e hiperintensas en T2 que pueden comprometer tanto el tronco encefálico como los ganglios de la base, el tálamo, cuerpo calloso, pedúnculo cerebeloso superior y cerebelo(3,13).
A nuestro paciente la RNM se le realizó en el día 5º de enfermedad y evidenció cambios parcheados y difusos de la señal en ganglios basales bilaterales, ambos pies de pedúnculos cerebrales, pedúnculo cerebeloso medio y regiones dorsales y antero laterales del bulbo raquídeo bilateral, coincidiendo con la literatura.
La presencia de anticuerpos anti GQ1b es de alta sensibilidad y especificidad para confirmar el diagnóstico en los Centros donde fuera posible su realización(1,3,4,5,7).
Repetimos que en el caso que hemos presentado no se ha realizado esta investigación por falta de disponibilidad laboratorio para investigaciones de esta complejidad.
El estudio del LCR en el SMF es posible encontrar una disociación albumino-citológica en cerca del 50% de los pacientes en la primera semana de instalado los síntomas y en un 80% luego de dos semanas, con un valor promedio de las proteínas de 62mg/dl, también se describe la presencia de disociación en la EB más o menos con la misma frecuencia(7,13,17). En nuestro caso la punción lumbar se realizó al sexto día de iniciado los síntomas y el resultado fue de cero células con 13 mg/dl de proteínas, es decir no hubo disociación albumino-citológica en este lapso.
Los hallazgos electrofisiológicos más consistentes en el SMF son la disminución de la amplitud de los potenciales de acción de nervios sensitivos con prolongación de las latencias distales o disminución de la velocidad de conducción sensitiva y la ausencia de la onda F y del reflejo H (4,7). Estudio de valor para los casos de SMF superpuesto a la EB.
En el caso que presentamos el resultado de la electromiografía y velocidad de conducción estuvo incompleto al no hacer referencia a los nervios sensitivos, ni al reflejo H. Se describió sin embargo una ligera disminución de la velocidad de conducción motriz y la onda F estuvo presente a 28 ms de latencia.
Los otros estudios complementarios que contribuyen al diagnóstico son los potenciales evocados auditivo del tronco cerebral (PEATC)y el Electroencefalograma(1,2,5).
La historia natural de la enfermedad es buena en la mayoría de los pacientes pediátricos como adultos. La recuperación completa se logra aun sin intervención. Se describe una recuperación espontánea en el 66% de los casos, usándose medidas de sostén para la fase aguda(3,9,26). En los niños pese a la poca evidencia, probablemente la IGIV acelere la recuperación comparado con las medidas de soporte solo. Más investigaciones serán necesarias para los casos de enfermedad leve o para aquellos cuyo tratamiento se inicien después de la segunda semana de enfermedad(26).
En nuestro caso el paciente recibió IGIV en el día 11 de enfermedad y podría servir de argumento para la rápida recuperación observada en su evolución, logrando su recuperación total a los 60 días.
Esto es así desde las descripciones históricas de este desorden(3-6,9,19). No obstante para las formas de presentación grave el tratamiento va dirigido al trastorno autoinmune con inmunoglobulinas, plasmaféresis o la combinación de ambos(1,4,9,12,13,21,26,27).
A pesar de los avances la Encefalitis de Bickerstaff sigue siendo objeto de controversias y de investigaciones(1).
CONCLUSION
El caso que se ha presentado cumple los criterios clínicos típicos para una Encefalitis del tallo cerebral de Bickersatff: cuadro con progresión dentro de las cuatro semanas caracterizado por ataxia, oftalmoplejía relativamente simétrica y alteración de la conciencia o signos piramidales. Estos síntomas de presentación aguda a subaguda deben tenerse presente para el diagnóstico sobre todo cuando se instalan después de una patología infecciosa respiratoria, gastrointestinal, o ambas. Es un cuadro poco frecuente en la infancia, pero puede ser grave. Por lo general la evolución es buena y la recuperación funcional es completa. La RNM cerebral fue relevante para sustentar el diagnóstico.
Claramente es una entidad que comparte rasgos clínicos con el síndrome de Miller Fisher y el síndrome de Guillain Barré, entidades de las cuales, muchas veces, es muy difícil de diferenciarlos. Debe tenerse una alta sospecha clínica, un buen examen físico y la implementación de los métodos auxiliares tales como la punción lumbar, la RNM cerebral y medular, la electromiografía, los potenciales evocados del tallo cerebral y el electroencefalograma. Es importante señalar que el estudio de anticuerpos antigangliósidos es un tema interesante para la investigación biomolecular.
REFERENCIAS
1. Lizarazo J, Jiménez F. Brainstem encephalitis of Bickerstaff. Acta Neurol Colomb. 2006;22:304-309. [ Links ]
2. Shahrizaila N, Yuki N. Bickerstaff brainestem encephalitis and Fisher síndrome: anti-Gq1b antibody syndrome. J Nneurol Neurosurg Psychiatry. 2013;84:576-83. [ Links ]
3. Susuki K, Atsumi M, Koga M, Hirata K, Yuki N. Acute facial diplegia and hyperreflexia: a Guillain-Barré síndrome variant. Neurology. 2004;62:825-27. [ Links ]
4. Pérez JC, Mateus SA, Mosquera JM. Encefalitis de Bickerstaff, syndrome o espectro de Fisher Bickerstaff, reporte de dos casos. Univ Med Bogotá (Colombia). 2011;52(3):315-24. [ Links ]
5. Park JY, Ko KO, Lim JW, Cheon EJ, Yoon JM, Kim HJ. A pediatric case of Bickerstaff s brainstem encephalitis. Korean J Pediatr. 2014;57(12):542-45. [ Links ]
6. Labbé F, López N, Mella D, López E. Síndrome de Miller Fisher: a propósito de un caso clínico. Revista Anacem. 2011;5(2):120-22. [ Links ]
7. Deschle F, Di Pace JL, Carnero Contentti E, Hryb JP, Perassolo M. Síndrome de Fisher- Bickerstaff; espectro clínico del anti-GQ1b. Neurol Arg. 2013;5(4). [ Links ] Doi: http://dx.doi.org/10.1016/j.neuarg.2013.08.003<
8. Lule-Alatorre KP, Domínguez-Borgua A, Martín-Ramírez JF, López-Galicia DN, Vázquez-Flores AD, Zaldivar-Clavellina AK. Bickerstaff: encephalitis del tallo cerebral. Med Int Mex. 2014;30(5):575-83. [ Links ]
9. Overell JR, Hsieh ST, Odaka M, Yuki N, Willison HJ. Treatment for Fisher Syndrome, Bickerstaff´s brainstem encephalitis and related disorders. Cochrane Database Syst Rev. 2007 Jan 24;(1):CD004761. [ Links ]
10. Kusunoki S, Chiba A, Kanazawa I. Anti- GQ1b IgG antibody is associated with ataxia as well as ophthalmoplegía. Muscle Nerve. 1999;22:1071-74. [ Links ]
11. Fargas i Busquets A, Roig Quils M, Gratacòs Vinyola M, Macaya Ruiz A, del Toro Riera M, Fitó Costa A. Oftalmoplejía-ataxia-arreflexia en pediatría: tres nuevos pacientes y revisión de la literatura. An EspPediatr. 1998;48(5):483-88. [ Links ]
12. Orphanet, the portal for rare disease and orphan drugs [Internet]. Encefalitis tronco encefálica de bickerstaff. París: Orphanet. [ Links ] Disponible en: http://www.orpha.net/consor/cgi-bin/index.php?Ing= Es
13. Guerra C, Uribe CS, Guerra A, Hernández OH. Encefalitis de Bickerstaff: informe de caso y revisión de la literatura. Biomédica. 2013;33(4):513-18. [ Links ]
14. Damasceno A, Franca Jr MC, Pimenta DS, de Deus-Silva L, Nucci A, Damasceno BP. Bickerstaff s encephalitis, Guillain-Barré syndrome and idiophaticintracraneal hypertension: are they related conditions?. Arq Neuropsiquiat. 2008;66(3-B):744-46. [ Links ]
15. Torricelli RE. Síndrome de Guillain Barré en pediatría. Medicina (Buenos Aires). 2009;69(1/1):84-91. [ Links ]
16. Lo YL. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve. 2007;36:615-27. [ Links ]
17. Odaka M, Yuki N, Yamada M, Koga M, Takemi T, Hirata K, Kuwabara S. Bickerstaff´s brainstem encephalitis clinical features of 62 cases and a subgroup associated with GuillainBarré Syndrome. Brain. 2003;126(Pt 10):2279-90. [ Links ]
18. Pan L, Yuki N, Koga M, Chiang MC, Hsieh ST. Acute sensory ataxic neuropathy associated with monospecific anti-GD1b IgG antibody. Neurology. 2001;57:1316-18. [ Links ]
19. Shahrizaila N, Yuki N. Guillain-Barré syndrome, Fisher syndrome and Bickerstaff brainstem encephalitis: understanding the pathogenesis. Neurology Asia. 2010;15(3):203-209. [ Links ]
20. Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Ganglioside composition of the human cranial nerves, with special reference to pathophysiology of Miller Fisher syndrome. Brain Res. 1997;745:32-36. [ Links ]
21. Hunter G, Young GB, Ang LC. Bickerstaff s brainstem encephalitis: presenting to the ICU. Neurocritical Care. 2012;17(1):102-106. [ Links ]
22. Yuki N, Hartung Hans P. Guillain-Barré syndrome. N Engel J Med. 2012;366:2294-2304. [ Links ]
23. Tolosa AC, Cartier RL. Infección por mycoplasmaneumoniae: microangeitis cerebral, síndrome de Bickerstaff, y anemia hemolítica autoinmune. Rev Chil Neuropsiquiat. 2011;49(1):56-61. [ Links ]
24. Pérez JC, Mateús SA, Mosquera JM. Encefalitis de Bickerstaff, syndrome o espectro de Fisher Bickerstaff: reporte de casos. Univ Med Bogotá (Colombia). 2011;52(3):315-24. [ Links ]
25. Ogawara K, Kuwabara S, Yuki N. Fisher syndrome or Bickerstaff brainstem encephalitis?: anti- GQ1b IgG antibody syndrome involving both the peripheral and central nervous systems. Muscle Nerve. 2002;26(6):845-49. [ Links ]
26. Hughes RA, Raphael JC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2006 Jan 25;(1):CD002063. [ Links ]
27. Ito M, Kuwabara S, Odaka M, Misawa S, Koga M, Hirata K, Yuki N. Bickerstaff s brainstem encephalitis and Fisher syndrome form a continuous spectrum: clinical analysis of 581 cases. J Neurol. 2008;255:674-82. [ Links ]

