










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPediatría (Asunción)
On-line version ISSN 1683-9803
Pediatr. (Asunción) vol.43 no.2 Asunción Aug. 2016
https://doi.org/10.18004/ped.2016.agosto.115-122
ARTÍCULO ORIGINAL
Diversidad de adenovirus detectados en niños menores de 5 años hospitalizados por infección respiratoria aguda baja en Paraguay, 2010-2013
Diversity of adenoviruses detected in children less than 5 years of age hospitalized with lower acute respiratory infection in Paraguay, 2010-2013
Julio C. Barrios(1), Graciela Russomando(1), Emilio E. Espínola(1)
1 Departamento de Biología Molecular y Biotecnología, Instituto de Investigación en Ciencias de la Salud, Universidad Nacional de Asunción. San Lorenzo, Paraguay.
Correspondencia: Emilio E. Espínola. Campus Universitario de la UNA; Dr. Cecilio Báez casi Dr. Gaspar Villamayor, CP 2160, San Lorenzo, Paraguay. E-mail: emilioespinola@hotmail.com
Conflicto de intereses: Los autores declaran no poseer conflicto de intereses.
Recibido: 16/06/2016; Aceptado: 18/07/2016.
RESUMEN
Los adenovirus humanos (HAdV) causan una diversidad de patologías que pueden ser leves o incluso mortales en pacientes vulnerables. Su evolución ocurre conforme actúa una presión de selección; por ejemplo, la recombinación genética genera cepas híbridas que pueden ser más infectivas o más patogénicas que las parentales. Este estudio buscó determinar la diversidad de HAdV circulantes en niños paraguayos menores de 5 años hospitalizados por infección respiratoria aguda baja, mediante el uso de herramientas de biología molecular y bioinformática, para lo cual fueron amplificados los genes hexon, penton y fibra en 26 muestras de aspirados nasofaríngeos. Se demostró la circulación de tres especies: B (4/26), C (21/26), y D (1/26), así como también eventos de recombinación en el aislado correspondiente a HAdV-D, al menos con tres genotipos distintos (D49, D9 y D15). Los HAdV-D están asociados a enfermedades gastrointestinales, oculares y, menos comúnmente a infecciones respiratorias. Sin embargo, recientemente se han reportado cambios en el comportamiento de estos virus debido a recombinación, lo que les permite expandir su repertorio de células huésped. Estos datos amplían los conocimientos acerca de la diversidad genética de HAdV en el Paraguay y apoyan fuertemente la importancia de los análisis genómicos mediante herramientas bioinformáticas en la vigilancia epidemiológica de las cepas recombinantes emergentes de HAdV.
Palabras clave: Infección respiratoria aguda baja, adenovirus, secuenciación ADN, análisis bioinformático, recombinación.
ABSTRACT
Adenoviruses cause a variety of diseases that range from mild to fatal in vulnerable patients. Its evolution occurs as a process of selection pressure; for example, genetic recombination generates intermediate strains that can be more infectious or pathogenic than parental strains. This study sought to determine the diversity of circulating adenoviruses in Paraguayan children less than 5 years of age hospitalized with acute lower respiratory infection, by using molecular biology and bioinformatics tools, though amplification of penton, hexon and fiber genes in 26 samples of nasopharyngeal aspirates. It was demonstrated the circulation of three species: B (4/26), C (21/26), and D (1/26), as well as recombination events corresponding to the HAdV-D isolate, with at least three different genotypes (D49, D9 y D15). The HAdV-D are associated with gastrointestinal or ocular diseases, and less commonly with respiratory infections. Recently, however, it has been reported changes in behavior of these viruses due to recombination, allowing them to expand their host cell repertoire. These data extend the knowledge about the genetic diversity of HAdV in Paraguay and strongly support the importance of genomic analysis using bioinformatics tools in epidemiological surveillance of emerging recombinant strains of HAdV.
Keywords: Acute lower respiratory tract infection, adenovirus, DNA sequencing, bioinformatic analyses, recombination.
INTRODUCCIÓN
Los adenovirus humanos (HAdV) pertenecen a la familia Adenoviridae, y poseen un genoma de DNA lineal de 34 a 36 kbp (1,2). Son una causa principal de infecciones respiratorias agudas bajas, principalmente en niños. Además, pueden comprometer los sistemas gastrointestinal, ocular, renal, entre otros, con billones de personas infectadas a nivel mundial(3). Comprenden 65 genotipos (HAdV-1 a HAdV-65) (4,5), de los cuales los primeros 51 tipos fueron caracterizados en base al tipo específico de neutralización mediada por antisuero(6,7) y los demás fueron designados por análisis bioinformático de secuencias(4). Los genotipos se agrupan en siete especies, denominadas HAdV-A a HAdV-G(8).
Los HAdV son virus no envueltos que presentan una cápside icosaédrica, con fibras que sobresalen de los vértices. La cápside rodea y protege el genoma del virus (1,2). Está constituida por 240 capsómeros de la proteína hexon, que forma la mayor parte de la superficie de la cápside, y contiene dos bucles hiper variables (bucle 1 y bucle 2) donde se encuentra el determinante antigénico Ɛ contra la cual reaccionan los anticuerpos en una reacción de neutralización (9). La cápside también posee 12 capsómeros de la proteína penton, uno en cada vértice (10); esta proteína tiene un motivo conservado de Arginina-Glicina-Ácido aspártico (RGD) que interactúa con las integrinas celulares y es importante en la internalización del virus en la célula huésped (11). A partir de cada penton sobresalen unas prolongaciones proteicas denominadas fibra, que se componen de tres dominios: un dominio N-terminal (tail) que se une a la base penton, un eje central (shaft) que presenta flexibilidad durante la infección y una porción C-terminal globular (knob) que se une al receptor primario en células huésped (12).
Entre los principales mecanismos de evolución descriptos para HAdV se encuentran las mutaciones puntuales y eventos de recombinación genética intra- o inter-especies (13). Estas últimas se desarrollan cuando dos o más genotipos diferentes de HAdV infectan a una misma célula y, eventualmente, la DNA polimerasa que realiza la replicación del HAdV donador pasa a replicar al HAdV aceptor, generando un genoma recombinante. Se ha reportado que los genes hipervariables hexon, penton y fibra son sitios altamente susceptibles a eventos de recombinación, ya sea en regiones intragénicas, o en sus zonas flanqueantes(14). No se conoce el mecanismo exacto de recombinación, sin embargo, algunos autores sugieren que la recombinación puede ser consecuencia de la presión selectiva del sistema inmune del huésped sobre la superficie proteica de la cápside viral, o un motivo de nucleótidos que dirige la maquinaria de recombinación celular hacia sitios específicos del ADN viral; o una combinación de ambas (15).
En Paraguay, uno de los pocos trabajos de detección de variabilidad genética de HAdV fue publicado en el año 2012, en el cual se analizaron muestras de aspirados nasofaríngeos (ANFs) de niños menores de 5 años hospitalizados por IRAb, detectándose la circulación de las especies: B (HAdV-16), C (HAdV-1, HAdV-2, HAdV-5, HAdV-6) y D (HAdV-15); los genotipos fueron designados en base al análisis filogenético de la región C4 del gen hexon (16).
En el presente trabajo se analizó la variabilidad de los 3 genes reportados como sitios probables de recombinación, es decir, el hexon, penton y fibra, utilizando como punto de partida las muestras paraguayas analizadas anteriormente sólo para el gen hexon (16), adicionando muestras colectadas durante el desarrollo de este trabajo y realizando un análisis bioinformático más profundo con el fin de detectar nuevos genotipos y posibles recombinantes inter-génicos de HAdV.
METODOLOGÍA
Muestras y extracción de DNA viral
Se analizaron 26 muestras de ANFs de niños menores de 5 años, hospitalizados con diagnóstico de IRAb en el Hospital General Pediátrico Niños de Acosta Ñu (San Lorenzo – Departamento Central, Paraguay), 2010-2013, en los cuales fueron detectados previamente HAdV por la técnica de PCR en tiempo real. El DNA viral se extrajo a partir de 200 µL de ANF, utilizando el AxyPrep Body Fluid Viral DNA/RNA Miniprep Kit (Axygen Biosciences, USA), y se eluyó en 60 µL de agua.
PCR convencional
Se amplificaron las regiones genómicas correspondientes a los genes hexon, penton y fibra. Se emplearon cebadores ya publicados para amplificar los genes hexon (17) y penton (18). Sin embargo, fue necesario el diseño de nuevos cebadores para el gen fibra de cada especie de HAdV, debido a la dificultad de amplificar este gen en muestras clínicas con los cebadores disponibles. En cuanto al gen hexon, se amplificó la región C4, empleando dos pares de cebadores (17), que amplifican de forma genérica todas las especies de HAdV sin distinción de genotipos. En cuanto al gen penton, se amplificó la región que contiene el motivo RGD, utilizando tres pares de cebadores (18), un par para cada especie (B, C y D). En cuanto al gen fibra, se amplificó un fragmento que incluía las regiones shaft/knob (en cuyos límites se han reportado eventos de recombinación); se diseñaron los cebadores forward para las especies B y D, y se emparejaron con cebadores descritos previamente (9). Para la especie C, se diseñaron tres cebadores genotipo específicos: un cebador forward (FiADV56-F) para los genotipos 1, 2, 5 y 6; un cebador reverse (FiADV12-R) para los genotipos 1 y 2 y otro reverse (FiADV56-R) para los genotipos 5 y 6. Las mezclas de reacción de PCR, condiciones de termociclado y electroforesis de los amplicones obtenidos, se realizaron según reportes publicados en esta sección.
Secuenciación, anotaciones y alineamientos múltiples de secuencias
La secuenciación directa se realizó en la empresa Macrogen Inc. (Corea), por el método de Sanger. Para la edición manual y anotaciones de secuencias, se utilizó el programa BioEdit 7.2.5 (19). Con cada anotación hecha se formaron tres matrices nucleotídicas distintas, una para cada gen. Estas matrices fueron sometidas a alineamientos múltiples utilizando Clustal W (20).
Construcción del árbol filogenético para los genes hexon, penton y fibra
Se realizó para los genes penton, hexon y fibra por separado para evaluar la coherencia de genotipos en cada cepa y detectar posibles recombinantes inter-génicos, utilizando el método de neighbor-joining, y K2P como modelo de sustitución nucleotídica, con valor de bootstrap de 1000 réplicas, utilizando el paquete de programas MEGA 5 (21). Los genotipos fueron asignados según su agrupamiento con secuencias de referencia extraídas de la base de datos GenBank.
Análisis de recombinación
Se utilizó el programa Simplot v 3.5.1 (22), utilizando un ancho de ventana (window size) de 200 nucleótidos y una longitud de movimiento de la ventana (step size) de 20 nucleótidos.
Aislamiento de virus en cultivo celular
Se procedió al aislamiento de HAdV en cultivo celular, utilizando la línea celular A549. Las muestras analizadas fueron seleccionadas por su naturaleza recombinante, y aquellas cuyos volúmenes fueron agotándose a medida que se estandarizaban la PCRs, para proseguir con los análisis.
Notación
Los genotipos resultantes fueron expresados utilizando letras mayúsculas para indicar el gen, seguido del genotipo asignado, como sigue: “P” para penton, “H” para hexon y “F” para fibra. Así P1H1F1 representa un virus “no recombinante” de genotipo 1 en los tres genes analizados. En caso de no determinarse el genotipo para un gen, se utilizó un símbolo de interrogación (ej: P?H1F1).
RESULTADOS
Genes amplificados mediante PCR convencional
De las 26 muestras clínicas positivas para HAdV, se logró amplificar los tres genes en diferente número de muestras: hexon (n=26), penton (n=22), y fibra (n=15) (Tabla 1).
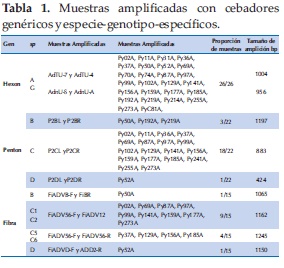
La no amplificación de algunos genes penton y fibra se debió a la escasez de muestra clínica (o crecimiento infructuoso en cultivo celular), o problemas de anillamiento de los cebadores disponibles. Cabe destacar que la amplificación de todos los genes hexon, en primer lugar, permitió la agrupación de las muestras por especies y la aplicación de cebadores especie-genotipo-específicos para los demás genes.
Filogenia del gen penton - Especie B
Para la especie B, la matriz se compuso de 3 secuencias en estudio y 57 secuencias de referencia (genotipos 1 al 57). Una se agrupó con HAdV-B16, y dos se asociaron con HAdV-B3 (Figura 1A).
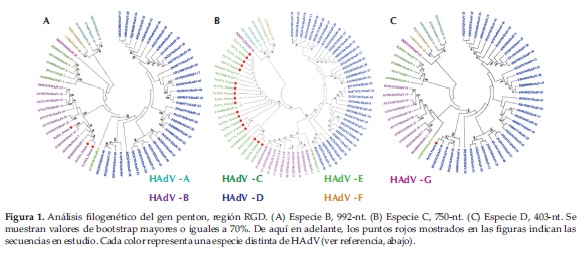
Para la especie C, la matriz se compuso de 18 secuencias en estudio y 57 secuencias de referencia. De las 18, cinco se agruparon con HAdV-C1, tres se agruparon con HAdV-C2, una con HAdV-C6, seis ocuparon una posición intermedia entre los genotipos de referencia HAdV-C1 y HAdV-C6, y tres ocuparon una posición intermedia entre los genotipos HAdV-C5 y HAdV-C6 (Figura 1B).
Para la especie D, la matriz se conformó con una secuencia en estudio y 57 secuencias de referencia. Py52A se agrupó con HAdV-D49 (Figura 1C).
Filogenia del gen hexon
Para la especie B, se obtuvieron 4 secuencias: tres de ellas se asociaron con HAdV-B3, y una se asoció previamente con HAdV-B16 (16) (Figura 2).
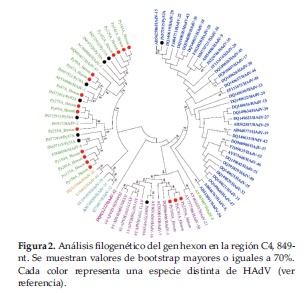
Para la especie C, se obtuvieron 21 secuencias: cinco de ellas se agruparon con HAdV-C1, cuatro con HAdV-C2, seis con HAdV-C5 y otras seis con HAdV-C6. De todas ellas, las marcadas con puntos rojos fueron nuevas determinaciones (17 secuencias) y las marcadas con puntos negros, ya fueron reportadas previamente (16) (Figura 2).
Una sola secuencia en estudio (Py52A) fue agrupada dentro de la especie D; esta secuencia fue previamente reportada como HAdV-D15 (16) (Figura 2).
Filogenia del gen fibra
Para la filogenia del gen fibra, se crearon dos matrices debido a diferencias en longitudes de nucleótidos secuenciados para la especie B con respecto a las especies C y D. Para la especie B, la matriz se conformó con una secuencia en estudio (Py50A) y 57 secuencias de referencia del mismo gen. Py50A se asoció con a HAdV-B16 (Figura 3A).
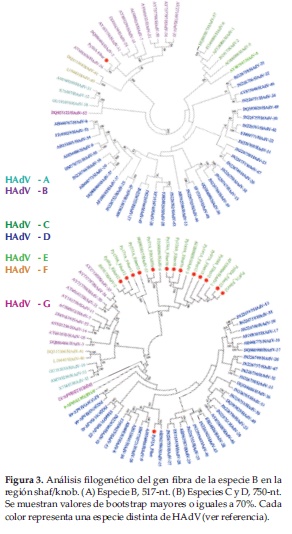
En cuanto a las especies C y D, la matriz incluyó 14 secuencias en estudio y 57 secuencias de referencia. Cinco se agruparon con HAdV-C1, cuatro con HAdV-C2, tres con HAdV-C5, y una con HAdV-C6. Py52A (único representante de la especie D), se agrupó con HAdV-D15 (Figura 3).
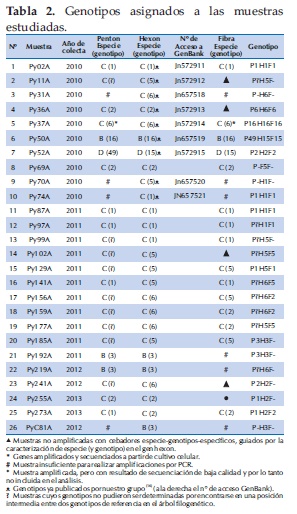
Genotipos asignados a las muestras en estudio
Una vez asignados los genotipos, Tabla 2, se observaron muestras cuyos genes pertenecían al mismo genotipo (es decir, probables no-recombinantes) y muestras con genotipos discordantes en uno o más genes (lo que indicaría la posible existencia de eventos de recombinación inter-génica en estas muestras). Por ejemplo, un probable no-recombinante sería la muestra N° 1 (Py02A), de genotipo 1 en los tres genes estudiados (P1H1F1); por otro lado, un ejemplo de probable recombinante inter-génico sería la muestra N° 7 (Py52A), con al menos dos genotipos diferentes (P49H15F15).
Análisis de recombinación
No se detectó evidencia contundente de recombinación intra-génicas en las regiones parciales de los genes analizados (datos no mostrados). Debido a estos resultados, se decidió extender el estudio con una de las muestras más representativas de este trabajo: Py52A (probable recombinante inter-génico, tipificado como P49H15F15) perteneciente a la especie D, cuyos miembros son poco frecuentes en infecciones respiratorias.
Análisis de recombinación para la especie D
En un intento de aclarar la inconsistencia genotípica observada anteriormente con la muestra Py52A, se aisló el virus a partir de cultivo celular, y fueron diseñados nuevos cebadores para amplificar una región de 1572-nt del gen hexon incluyendo parte del bucle 1, que contiene 6 regiones hiper variables (HVR1-6), y el bucle 2, que contiene la HVR-7, donde comúnmente son reportados eventos de recombinación intra-génica (23). Este fragmento de 1572-nt fue empalmado a la región C4 anteriormente obtenida, resultando en un fragmento final de 2370-nt (renombrado Py52A-HexCult). El análisis filogenético de esta región mostró la asociación de Py52A-HexCult con HAdV-9 (Figura 4).
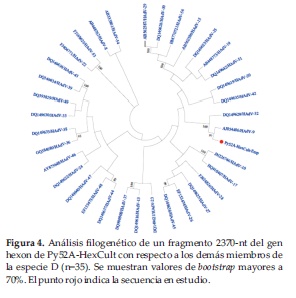
Debido a que esta asociación con HAdV-9 fue inesperada, la matriz fue sometida a análisis de recombinación. El análisis Simplot demostró que los bucles 1 y 2 del gen hexon presentaban más del 95% de similitud con HAdV-9, mientras que la región C4 del gen hexon presentó más del 98% de similitud con HAdV-15, sugiriendo un evento de recombinación entre éstos dos prototipos (Figura 5).
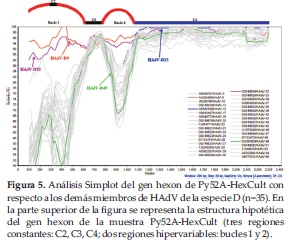
Para encontrar el punto exacto de quiebre o recombinación se realizó un análisis bootscan (valores por defecto, datos no mostrados), mediante el cual se detectó un evento de recombinación al final del bucle 2 e inicio de la región C4, en la posición nucleotídica 1100, aproximadamente.
DISCUSIÓN
En este estudio se analizaron 26 muestras de ANFs positivas para HAdV, provenientes de pacientes menores de 5 años hospitalizados por IRAb, con la finalidad de determinar la diversidad genética de estos virus en Paraguay y la presencia de eventos de recombinación.
Las especies de HAdV estudiadas aquí y descriptas concuerdan con las reportadas previamente en nuestro país (16); sin embargo, se reporta el hallazgo de nuevos genotipos (por ejemplo, HAdV-B3), la detección de cepas puras teniendo en cuenta las tres regiones analizadas (P1H1F1, P2H2F2, P6H6F6, P16H16F16) y la confirmación de cepas recombinantes intra-génicos (P49H9/15F15) e inter-génicos (P1H2F2, P1H5F1, entre otros).
Las asignaciones de genotipos fueron provisorias, ya que se analizaron secuencias parciales de cada gen; en el gen hexon, por ejemplo, se amplificó la porción terminal de la región C4, caracterizada por ser altamente conservada en todos los adenovirus; en la especie C, ésta región es indistinguible entre los prototipos por la alta similitud entre ellas y por presentar en ella eventos de recombinación. De esta forma, el prototipo HAdV-C6 contiene un fragmento HAdV-C2 en ésta región (C6/C2) (24). Para establecer un genotipo definitivo a las muestras, se deberá secuenciar otras regiones del gen hexon como los bucles 1 y 2, que presentan variabilidad para distinguir un genotipo de otro (25).
Algunos genes de HAdV fueron agrupados en una posición intermedia entre dos genotipos distintos (ej. para el gen penton de la especie C, entre genotipos HAdV-C1 y HAdV-C6). Sin embargo, fueron descartados como recombinantes, teniendo en cuenta las regiones analizadas. En estos casos, para confirmar/descartar recombinación, se deberá amplificar el gen penton completo. Otros reportes ya han demostrado la ocurrencia de recombinación en el penton de otras especies, ocasionando un cambio del tropismo celular (15).
En cuanto a los recombinantes inter-génicos, se observaron varios ejemplos, tales como P1H2F2, o P1H5F1. No se descarta la probabilidad de que estos sean genomas completamente nuevos, ya que la recombinación dentro de la especie C es más común de lo que se pensaba, ejemplo de ello es el recombinante HAdV-C57 (P1H57F6) (24,26).
Los primeros análisis de Py52A indicaron que se trataba de un recombinante inter-génico conteniendo el penton del genotipo HAdV-D49 y el hexon y fibra del genotipo HAdV-D15 (P49H15F15). La idea de encontrar el punto de quiebre entre el genotipo -D49 y el -D15 en el gen hexon, nos llevó a amplificar la mayor parte de este gen. Los análisis realizados nos indicaron que se trataba de un genoma con múltiples eventos de recombinación con al menos tres genotipos: (i) gen penton del genotipo -D49; (ii) gen hexon quimérico, con genotipos -D9 y -D15; (iii) gen fibra del genotipo –D15. Por lo tanto, el aislado Py52A cuyo genotipo se designó finalmente como P49H9F15, sería un nuevo genotipo de HAdV.
En el Paraguay, este trabajo es el segundo en abordar la diversidad genética de HAdV y el primero en demostrar la naturaleza recombinante de cepas aisladas de niños menores de 5 años con IRAb. Además, demuestra la importancia de seguir profundizando en la epidemiología molecular de adenovirus en el país, debido a que en cualquier momento podría surgir una cepa recombinante altamente patogénica de importancia en salud pública.
REFERENCIAS
1. Benko M, Harrach B, Russell WC. Family adenoviridae. In: van Regenmortel HV, Ed. Virus taxonomy: seventh report of the International Committee on Taxonomy of Viruses. San Diego: Academic Press; 2000. p. 227-38. [ Links ]
2. Rux JJ, Burnett RM. Adenovirus structure. Hum Gene Ther. 2004;15(12):1167-76. [ Links ]
3. Lynch JP, Fishbein M, Echavarria M. Adenovirus. Semin Respir Crit Care Med. 2011;32(4):494-511. [ Links ]
4. Robinson CM, Zhou X, Rajaiya J, Yousuf MA, Singh G, DeSerres JJ, Walsh MP, Wong S, Seto D, Dyer DW, Chodosh J, Jones MS. Predicting the next eye pathogen: analysis of a novel adenovirus. MBio. 2013;4(2):e00595-12. [ Links ]
5. Matsushima Y, Shimizu H, Kano A, Nakajima E, Ishimaru Y, Dey SK, Watanabe Y, Adachi F, Mitani K, Fujimoto T, Phan TG, Ushijima H. Genome sequence of a novel virus of the species human adenovirus d associated with acute gastroenteritis. Genome Announc. 2013;1(1): e00068-12. [ Links ]
6. Hierholzer JC, Stone YO, Broderson JR. Antigenic relationships among the 47 human adenoviruses determined in reference horse antisera. Arch Virol. 1991;121(1-4):179-97. [ Links ]
7. De Jong JC, Wermenbol AG, Verweij-Uijterwaal MW, Slaterus KW, Wertheim-Van Dillen P, Van Doornum GJ, Khoo SH, Hierholzer JC. Adenoviruses from human immunodeficiency virus-infected individuals, including two strains that represent new candidate serotypes Ad50 and Ad51 of species B1 and D, respectively. J Clin Microbiol. 1999;37(12):3940-5. [ Links ]
8. Kajon AE, Moseley JM, Metzgar D, Huong HS, Wadleigh A, Ryan MA, Russell KL. Molecular epidemiology of adenovirus type 4 infections in US military recruits in the postvaccination era (1997-2003). J Infect Dis. 2007;196(1):67-75. [ Links ]
9. Madisch I, Harste G, Pommer H, Heim A. Phylogenetic analysis of the main neutralization and hemagglutination determinants of all human adenovirus prototypes as a basis for molecular classification and taxonomy. J Virol. 2005;79(24):15265-76. [ Links ]
10. Ginsberg HS, Pereira HG, Valentine RC, Wilcox WC. A proposed terminology for the adenovirus antigens and virion morphological subunits. Virology. 1966;28(4):782-3. [ Links ]
11. Zubieta C, Schoehn G, Chroboczek J, Cusack S. The structure of the human adenovirus 2 penton. Mol Cell. 2005;17(1):121-35. [ Links ]
12. Wu E, Pache L, Von Seggern DJ, Mullen TM, Mikyas Y, Stewart PL, Nemerow GR. Flexibility of the adenovirus fiber is required for efficient receptor interaction. J Virol. 2003;77(13):7225-35. [ Links ]
13. Walsh MP, Chintakuntlawar A, Robinson CM, Madisch I, Harrach B, Hudson NR, Schnurr D, Heim A, Chodosh J, Seto D, Jones MS. Evidence of molecular evolution driven by recombination events influencing tropism in a novel human adenovirus that causes epidemic keratoconjunctivitis. PLoS One. 2009;4(6):e5635. [ Links ]
14. Robinson CM, Singh G, Lee JY, Dehghan S, Rajaiya J, Liu EB, Yousuf MA, Betensky RA, Jones MS, Dyer DW, Seto D, Chodosh J. Molecular evolution of human adenoviruses. Sci Rep. 2013;3:1812. [ Links ]
15. Robinson CM, Rajaiya J, Walsh MP, Seto D, Dyer DW, Jones MS, Chodosh J. Computational analysis of human adenovirus type 22 provides evidence for recombination among species D human adenoviruses in the penton base gene. J Virol. 2009;83(17):8980-5. [ Links ]
16. Espinola EE, Russomando G, Basualdo W, Benítez DA, Meza G, Maldonado L, Paranhos-Baccalà G. Genetic diversity of human adenovirus in hospitalized children with severe acute lower respiratory infections in Paraguay. J Clin Virol. 2012;53(4):367-69. [ Links ]
17. Saitoh-Inagawa W, Oshima A, Aoki K, Isobe K, Uchio E, Ohno S, Nakajima H, Hata K, Ishiko H. Rapid diagnosis of adenoviral conjunctivitis by PCR and restriction fragment length polymorphism analysis. J Clin Microbiol. 1996;34(9):2113-6. [ Links ]
18. Madisch I, Hofmayer S, Moritz C, Grintzalis A, Hainmueller J, Pring-Akerblom P, Heim A. Phylogenetic analysis and structural predictions of human adenovirus penton proteins as a basis for tissue-specific adenovirus vector design. J Virol. 2007;81(15):8270-81. [ Links ]
19. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series. 1999;41:95-98. [ Links ]
20. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673-80. [ Links ]
21. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731-9. [ Links ]
22. Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1999;73(1):152-60. [ Links ]
23. Liu EB, Ferreyra L, Fischer SL, Pavan JV, Nates SV, Hudson NR, Tirado D, Dyer DW, Chodosh J, Seto D, Jones MS. Genetic analysis of a novel human adenovirus with a serologically unique hexon and a recombinant fiber gene. PLoS One. 2011;6(9):e24491. [ Links ]
24. Walsh MP, Seto J, Liu EB, Dehghan S, Hudson NR, Lukashev AN, Ivanova O, Chodosh J, Dyer DW, Jones MS, Seto D. Computational analysis of two species C human adenoviruses provides evidence of a novel virus. J Clin Microbiol. 2011;49(10):3482-90. [ Links ]
25. Gall JG, Crystal RG, Falck-Pedersen E. Construction and characterization of hexon-chimeric adenoviruses: specification of adenovirus serotype. J Virol. 1998;72(12):10260-4. [ Links ]
26. Lukashev AN, Ivanova OE, Eremeeva TP, Iggo RD. Evidence of frequent recombination among human adenoviruses. J Gen Virol. 2008;89(Pt 2):380-8. [ Links ]

