










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPediatría (Asunción)
On-line version ISSN 1683-9803
Pediatr. (Asunción) vol.42 no.2 Asunción Aug. 2015
https://doi.org/10.18004/ped.2015.agosto.115-119
ARTÍCULO ORIGINAL
Experiencia en el tratamiento de Enfermedad de Gaucher con Imiglucerasa en el departamento de Hemato-Oncología Pediátrica del Hospital de Clínicas
Experience in the treatment of Gaucher disease with Imiglucerase in the Department of Pediatric Hematology-Oncology at the University Hospital
Diego Figueredo(1), Ofelia Zelada(1), Jazmín Servín(1), Isabel Mattio(1), Angélica Samudio(1)
1. Departamento de Hemato-Oncología Pediátrica, Facultad de Ciencias Médicas, Universidad Nacional de Asunción. San Lorenzo, Paraguay.
Correspondencia: Dr. Diego Figueredo. E-mail: figueredodiego@hotmail.com
Recibido: 23/01/2015; Aceptado: 16/03/2014.
Los autores declaran que no existen conflictos de interés en el presente estudio.
RESUMEN
Introducción: La enfermedad de Gaucher (EG) es el trastorno metabólico de depósito más frecuente. Es el resultado de una mutación autosómica recesiva en el gen que codifica la enzima ß-glucosidasa ácida. La deficiencia enzimática produce la acumulación de la glucosilceramida en los lisosomas del sistema monocito macrófago llevando a la acumulación de glucocerebrósidos, causando daño celular y disfunción orgánica. Objetivos: Describir las características clínicas de pacientes diagnosticados con EG en el Departamento de Hemato-Oncología Pediátrica del Hospital de Clínicas y evaluar la respuesta terapéutica a la Terapia de Reemplazo Enzimático (TRE) con la enzima Imiglucerasa. Materiales y Métodos: Estudio descriptivo de corte transversal, realizado entre enero de 2005 y diciembre de 2013. Resultados: En el periodo de estudio fueron diagnosticados 8 pacientes, 5 de sexo femenino. La edad media al diagnóstico fue de 4,5 años. Todos presentaron esplenomegalia, 7/8 hepatomegalia y 5/8 tenían afectación ósea al diagnóstico. Todos presentaron afectación hematológica. Con la TRE 7/8 alcanzaron las metas terapéuticas. El tiempo medio de normalización de la hemoglobina fue de 5 meses, el de las plaquetas de 11 meses. El tamaño del hígado se normalizó en promedio a los 10 meses y el bazo a los 14 meses. Conclusiones: La EG presenta gran heterogeneidad clínica. La respuesta a la TRE es por lo general excelente, siendo fundamental una adecuada adherencia al tratamiento.
Palabras clave: Enfermedad de Gaucher, reemplazo enzimático, Imiglucerasa.
ABSTRACT
Introduction: Gaucher disease (GD) is the most common metabolic deposit disorder. It is the result of an autosomal recessive mutation in the gene encoding the glucocerebrosidase enzyme. This enzyme deficiency results in the accumulation of glucosylceramide in the lysosomes of the monocyte macrophage system, leading to the accumulation of glucocerebrosides, causing cellular damage and organ dysfunction. Objectives: To describe the clinical characteristics of patients diagnosed with GD in the Department of Pediatric Hematology-Oncology at University Hospital and evaluate the therapeutic response to Enzyme Replacement Therapy (ERT) with imiglucerase enzyme. Materials and Methods: Cross-sectional study, conducted between January 2005 and December 2013. Results: During the study period 8 patients were diagnosed, 5 of them females. The mean age at diagnosis was 4.5 years. All had splenomegaly, 7 of 8 had hepatomegaly and 5 of 8 had bone involvement at diagnosis. All had hematologic involvement. With ERT, 7 of 8 achieved therapeutic goals. The median time to normalization of hemoglobin was 5 months and for platelets, 11 months. The average time for liver size normalization was 10 months and for spleen size normalization, 14 months. Conclusions: GD has great clinical heterogeneity. The response to ERT is usually excellent, but proper adherence to treatment remains fundamental.
Keywords: Gaucher disease, enzyme replacement, Imiglucerase.
INTRODUCCIÓN
La enfermedad de Gaucher (EG) es el trastorno metabólico de depósito lisosomal más frecuente. Se trata de un padecimiento crónico, progresivo y multisistémico (1). La enfermedad es el resultado de una mutación autosómica recesiva en el gen que codifica la síntesis de la enzima lisosomal ß -glucosidasa ácida (ß-GA). El defecto genético se localiza en el cromosoma 1, en la región q2.1. La deficiencia enzimática produce la acumulación de la glucosilceramida en los lisosomas del sistema monocito macrófago (SMM) llevando a la acumulación de glucocerebrósidos en médula ósea, bazo, hígado, pulmones y en las formas neurológicas en cerebro, causando daño celular y disfunción orgánica (1).
El espectro clínico de la enfermedad es altamente heterogéneo, existiendo formas severas en la infancia y formas de severidad variable en la edad adulta. La prevalencia se estima en 1 por cada 100,000 personas en la población general. La población más propensa de resultar afectada son los judíos oriundos de Europa Central y Oriental (Azquenazi). Se estima que en Paraguay, con una población aproximada de 6 millones de habitantes, deberían existir aproximadamente 60 pacientes con esta patología.
Aunque la enfermedad de Gaucher se manifiesta a lo largo de un espectro, por lo general se clasifica clínicamente en tres subtipos: Tipo 1 (no neuropático), Tipo 2 (neuropático agudo), y el Tipo 3 (neuropático crónico). Se reconocen dos fenotipos principales: una forma no peuropática (EG tipo 1) y una forma neuropática (EG tipos 2 y 3) (1,2).
En Europa, Estados Unidos y Latino América, la forma más frecuente es la EG de tipo 1, que se caracteriza por la ausencia de afectación del sistema nervioso central primario. En otras partes del mundo, como Egipto y Japón, las formas neuropáticas pueden tener una prevalencia más altas.
Los síntomas más frecuentes son visceromegalias (hepato-esplenomegalias), dolores óseos y citopenias, también puede haber alguna manifestación neurológica, con síntomas como depresión, síndrome del túnel carpiano, síntomas de la enfermedad de Parkinson, neuropatía periférica, etc. (3-5).
El pronóstico de la EG cambió radicalmente desde la aparición de la terapia de reemplazo enzimática (TRE) con Imiglucerasa. La intervención terapéutica temprana con dosis adecuadas de TRE permite, especialmente en niños, evitar la aparición de complicaciones óseas irreversibles.
Las metas terapéuticas a alcanzar con la TRE se han establecido como unas guías para los médicos tratantes de pacientes con EG y son utilizadas como método de decisión para evaluar la respuesta a la TRE. Los parámetros evaluados son: Hemoglobina: Incrementar los niveles de hemoglobina en los primeros 12 a 24 meses a: más de 11 g/dl en niños y mayor a 12 g/dl en mayores a 12 años, eliminar el requerimiento transfusional, reducir la fatiga y/o disnea, mantener los valores de hemoglobina alcanzados luego de los primeros 12 a 24 meses. Contaje de Plaquetas: incrementar el recuento de plaquetas durante el primer año de TRE lo suficiente para prevenir hemorragias, el recuento plaquetario debe incrementarse 1,5 a 2 veces en el primer año y aproximarse al nivel normal en el segundo, mantener estable el recuento plaquetario para eliminar los riesgos de hemorragia luego de que se ha alcanzado una respuesta terapéutica. Para el Compromiso óseo: disminuir o eliminar el dolor óseo en los primeros 2 años de tratamiento, prevenir las crisis óseas. Visceromegalias: reducir y mantener el volumen hepático en 1 a 1,5 veces de lo normal, reducir el volumen hepático en un 20-30% en el primer y segundo año, y un 30-40% en los siguientes 3 a 5 años, reducir y mantener el volumen esplénico en < 2 a 8 veces su tamaño normal, reducir el volumen esplénico en un 30-50% en el primer año y 50-60% en los siguientes 2 a 5 años, aliviar los síntomas por la esplenomegalia, eliminar el hiperesplenismo. La adherencia a la TRE es fundamental para el logro de las metas terapéuticas y evitar los daños óseos severos (6,7).
Este estudio tiene por objetivo describir las características clínicas de pacientes diagnosticados con Enfermedad de Gaucher y evaluar la respuesta terapéutica a la Terapia de Reemplazo Enzimático (TRE) con Imiglucerasa en los pacientes con Enfermedad de Gaucher.
MATERIALES Y MÉTODOS
Estudio prospectivo, descriptivo de corte transversal de pacientes con Enfermedad de Gaucher diagnosticados en el Departamento de Hemato-Oncología Pediátrica del Hospital de Clínicas, entre enero de 2005 y diciembre de 2013.
Fueron incluidos pacientes pediátricos entre 0 y 18 años de edad con diagnóstico confirmado de Enfermedad de Gaucher por método enzimático que demuestre la actividad deficiente de la enzima β-GA. Fueron excluidos a aquellos pacientes que abandonaron el tratamiento.
Se recolectaron datos respecto al género, edad al diagnóstico, signos de presentación, edad al inicio de la terapia, respuesta al tratamiento.
Los valores de hemoglobina, glóbulos blancos y plaquetas fueron evaluados laboratorialmente en contadores hematimétricos automatizados (Cell-Dyn Ruby) y corroborados por médicos hematólogos del Departamento. Las visceromegalias fueron evaluadas por clínica y por ecografía abdominal. La enfermedad ósea fue evaluada por clínica (dolores óseos, crisis óseas) y por Radiografías óseas.
RESULTADOS
Características clínicas: En este periodo de tiempo fueron diagnosticados 8 pacientes con Enfermedad de Gaucher tipo I. Fueron 5 de sexo femenino y 3 de sexo masculino. La edad media al diagnóstico fue de 4,5 años (Rango: 1 y 11 años).
Presentaron afectación ósea al diagnóstico 5/8 pacientes. El tamaño promedio del hígado fue de 7 cm (Rango: 3 y 13 cm), el del bazo de 12,5 cm (Rango: 6,5 y 20 cm) (Tabla 1). Presentó hemorragia al momento del diagnóstico 1/8. Presentaron clínica de afectación ósea al momento de diagnóstico 5/8, presentaron hepatomegalia 7/8 y 8/8 presentaron esplenomegalia. Ninguno de los pacientes presentó afectación pulmonar o cardiaca.

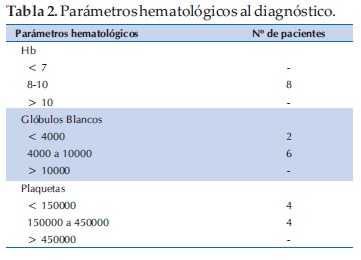
Estudios laboratoriales: En cuanto a los datos de laboratorio 8/8 presentaron afectación hematológica, que varió de una anemia leve a pancitopenia. La media de la Hb fue de 9,7 g/dl (Rango: 8 y 10,8 g/dl), de los GB 4520/mm3 (Rango: 3800 y 5200 mm3) y de las plaquetas 127.000 (Rango: 38.000 y 200.000) (Tabla 2).
Respuesta al tratamiento: El tiempo de seguimiento fue de 5 años (Rango 2 y 9 años), 7/8 alcanzaron las metas terapéuticas con la TRE. Los pacientes que alcanzaron las metas terapéuticas, el tiempo medio de recuperación de la Hemoglobina fue de 5 meses (Rango: 3 y 6 meses), el de las plaquetas de 11 meses (Rango: 3 y 18 meses).
Todos los pacientes lograron la remisión de los síntomas óseos con la terapia. El tamaño del hígado se normalizó a los 10 meses (Rango: 3 y 24 meses), el tamaño del bazo se normalizó a los 14 meses (Rango: 6 y 24 meses) (Figuras 1, 2,3, 4 y 5).
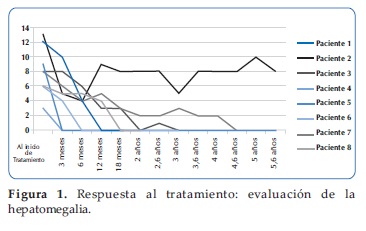
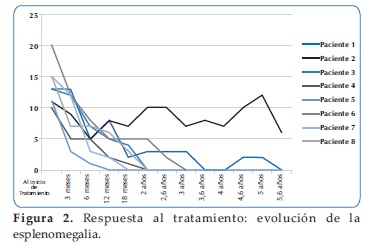


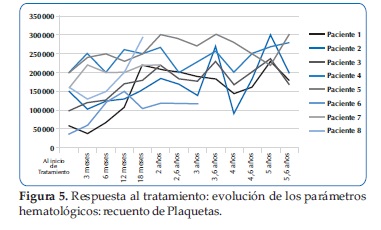
DISCUSIÓN
La enfermedad de Gaucher presenta gran heterogeneidad clínica. Pueden encontrarse pacientes desde muy severamente afectados hasta individuos asintomáticos. La afectación del bazo fue constante en todos los pacientes.
En nuestra serie de pacientes las metas terapéuticas fueron alcanzadas dentro del periodo de tiempo descripto por la literatura, uno sólo no alcanzó todas las metas terapéuticas, consideramos la baja adherencia al tratamiento como causa principal, tal como se describe en la literatura (8,9).
Observamos otros pacientes con comportamiento irregular, la paciente 1, hermana de la paciente 2, a pesar de la baja adherencia al tratamiento, logró las metas terapéuticas. La paciente 6 quien inicia el tratamiento con el menor recuento de plaquetas, sin bien no normalizó el recuento plaquetario en su tercer año de tratamiento logró las metas terapéuticas en todos los aspectos.
Debemos resaltar la importancia de realizar un adecuado seguimiento del compromiso óseo de todos los pacientes (10,11).
La respuesta al tratamiento de TRE con Imiglucerasa es por lo general excelente. El reconocimiento de la Enfermedad de Gaucher conduciría a un tratamiento seguro y eficaz con terapia de reemplazo enzimático lo que lleva a disminuir la morbilidad y reducir la afectación visceral y esquelética.
REFERENCIAS
1. Drelichman G, Basack N, Fernández Escobar N, Watman N, Bolesina M, Elena G, Veber SE, Kohan R, Dragosky M, Annetta I, Feliu A, Sciuccati G, Cuello MF, Fynn A, Dodelson de Kremer R, Angaroni CJ, Giner-Ayala AN, Oller de Ramírez A, Guelbert NB, Delgado MA, Becerra A, Oliveri B, Larroudé MS, Masllorens FM, Szlago M, Schenone AB, Aguilar G, Volpacchio M. Consenso para la Enfermedad de Gaucher: grupo argentino de diagnóstico y tratamiento de la Enfermedad de Gaucher. Hematología. 2013;17(Supl):25-60. [ Links ]
2. Kaplan P, Baris H, De Meirleir L, Di Rocco M, El-Beshlawy A, Huemer M, Martins AM, Nascu I, Rohrbach M, Steinbach L, Cohen IJ. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013;172(4):447-58. [ Links ]
3. Franco-Ornelas, S. Consenso Mexicano de Enfermedad de Gaucher. Rev Med Inst Mex Seguro Soc. 2010;48(2):167-86. [ Links ]
4. Charrow J, Dulisse B, Grabowski GA, Weinreb NJ. The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease. Clin Genet. 2007;71(3):205-211. [ Links ]
5. Balwani M, Fuerstman L, Kornreich R, Edelmann L, Desnick RJ. Type 1 Gaucher Disease: significant disease manifestations in asymptomatic homozygotes. Arch Intern Med. 2010;170(16):1463-69. [ Links ]
6. Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis. 2005;35(3):355-64. [ Links ]
7. Goldblatt J, Fletcher JM, McGill J, Szer J, Wilson M. Enzyme replacement therapy drug holiday: results from an unexpected shortage of an orphan drug supply in Australia. Blood Cells Mol Dis. 2011;46(1):107-110. [ Links ]
8. Pastores GM, Weinreb NJ, Aerts H, Andria G, Cox TM, Giralt M, Grabowski GA, Mistry PK, Tylki-Szymanska A. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 Suppl 5):4-14. [ Links ]
9. Mistry PK, Deegan P, Vellodi A, Cole JA, Yeh M, Weinreb NJ. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol. 2009;147(4):561-70. [ Links ]
10. Roca Espiau M. Aspectos óseos de la enfermedad de Gaucher. Med Clin (Barc). 2011;137(Supl 1):23-31. [ Links ]
11. Poll LW, Cox ML, Godehardt E, Steinhof V, vom Dahl S. Whole body MRI in type I Gaucher patients: evaluation of skeletal involvement. Blood Cells Mol Dis. 2011;46(1):53-59. [ Links ]

