










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPediatría (Asunción)
versão On-line ISSN 1683-9803
Pediatr. (Asunción) v.41 n.2 Asunción ago. 2014
CASO CLÍNICO
Síndrome de McCune-Albright. Reporte de un caso
McCune-Albright syndrome: a Case Report
Cynthia Florentín, Zoilo Morel, Raúl Gulino, Melva Galeano, Luis Chamorro, Fabiola Blanco(1)
1. Sala de Medicina Interna, Servicio de Pediatría, Hospital Central del Instituto de Previsión Social. Asunción, Paraguay.
Correspondencia: Dr. Zoilo Morel. Edificio Coomecipar. Piso 7. Asunción, Paraguay. E-mail: zoiloma@hotmail.com
Recibido: 30/12/2013; Aceptado: 28/02/2014.
RESUMEN
El Síndrome de McCune-Albright (SMA) es una rara entidad que se caracteriza por displasia fibrosa ósea poliostótica, lesiones cutáneas hiperpigmentadas y endocrinopatías, la más frecuente es la pubertad precoz y sobre todo en niñas. Presentamos el caso de una paciente de sexo femenino de 5 años de edad, que se interna por fractura patológica del fémur derecho, constatándose lesiones líticas en fémur contralateral, pelvis, tórax y calota; manchas café con leche en regiones del tórax anterior, perineal y dorsolumbar; Tanner 2 mamario y púbico, con antecedente de sangrado vaginal en 2 oportunidades 1 mes antes; y con Rx de muñeca izquierda compatible con edad ósea de 9 años; además de microadenoma hipofisiario. El SMA resulta de mutaciones esporádicas somáticas postcigóticas en el gen que codifica la subunidad α de la proteína Gs (GNAS1). Esta proteína actúa en la transducción de señales mediante la unión a la adenil-ciclasa productora de adenosín monofosfato cíclico (AMPc). Es importante conocer esta asociación de signos a fin de obtener un diagnóstico precoz y manejo adecuado.
Palabras clave: Síndrome de McCune-Albright, displasia fibrosa ósea, pubertad precoz, manchas café con leche, niños.
ABSTRACT
McCune-Albright syndrome (MAS) is a rare disease characterized by polyostotic fibrous dysplasia of bone, hyperpigmented skin lesions, and endocrinopathies, most commonly precocious puberty, and especially in girls. We presented the case of a female patient aged 5 years hospitalized for pathological fracture of the right femur with findings of lytic lesions of the contralateral femur, pelvis, thorax, and calvarium, and café-au-lait spots of the anterior, perineal, and dorsolumbar thorax; Tanner stage 2 breasts and pubes, a history of vaginal bleeding on two occasions one month earlier, left-wrist X-ray compatible with a bone age of 9 years and pituitary microadenoma. MAS is caused by sporadic postzygotic somatic mutations of the gene that codifies the alpha subunit of the G(s) protein (GNAS1). This protein acts in the transduction of signals by binding with cyclic-adenosine-monophosphate (cAMP) producing adenylate cyclase. It is important to be aware of this group of associated signs in order to achieve early diagnosis and appropriate treatment.
Keywords: McCune-Albright syndrome, fibrous dysplasia of bone, precocious puberty, café-au-lait spots, children.
INTRODUCCIÓN
El Síndrome de McCune-Albright está constituido por la tríada clásica de manchas café con leche, displasia fibrosa ósea y pubertad precoz. Para el diagnóstico, sólo son necesarias dos de estas tres alteraciones(1-3).
Se considera una entidad extremadamente rara, cuya incidencia es desconocida, y la prevalencia estimada oscila entre 1/100.000 y 1/1.000.000, siendo más frecuente en el sexo femenino(4). Se detecta a cualquier edad, sin distinción de raza, y cualquier hueso puede estar afectado. El SMA resulta de mutaciones esporádicas somáticas postcigóticas en el gen que codifica la subunidad α de la proteína Gs (GNAS1). Esta proteína actúa en la transducción de señales mediante la unión a la adenil-ciclasa productora de adenosín monofosfato cíclico (AMPc). La ocurrencia esporádica y la heterogenicidad de las manifestaciones clínicas, dependen del número de líneas celulares comprometidas que origina un mosaicismo para el gen mutado(5-8).
El AMPc intracelular, normalmente estimula la proliferación de las células en las glándulas (tiroides, corteza suprarrenal, ovario y pituitaria) que dan lugar a los tumores hiperfuncionantes en estos pacientes. Las manchas café con leche, únicas o múltiples, aparecen en el 2/3 de los casos. La hiperpigmentación cutánea no resulta de la proliferación excesiva de las células, se produce a partir de la imitación del efecto normal de la hormona estimulante de los melanocitos en los receptores que utilizan Gs para aumentar la síntesis de cAMP en los melanocitos. A diferencia de las manchas café con leche de la neurofibromatosis, en el SMA se presentan en número escaso, con bordes dentados y más grandes. Su distribución es característica, no rebasan la línea media y se localizan en el mismo lado de la afección ósea más grave, y son más frecuentes en sacro, glúteos y región lumbar.
Presentamos el caso de una paciente de sexo femenino con dicha patología.
CASO CLÍNICO
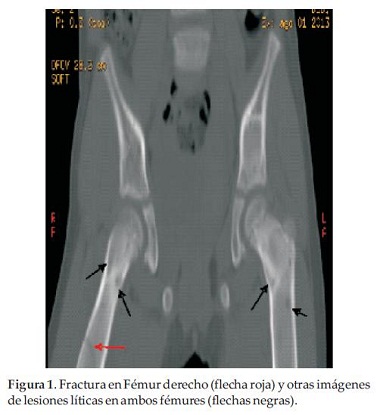
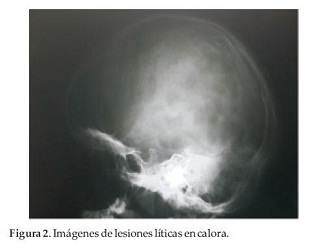
Paciente de sexo femenino de 5 años de edad, consulta por dolor de miembro inferior derecho de 48 horas de evolución, posterior a caída de propia altura, que no cede con analgésicos comunes, constatándose fractura patológica de fémur derecho e imágenes de lesiones líticas en fémur contralateral, pelvis, tórax y calota (Figura 1 y 2).
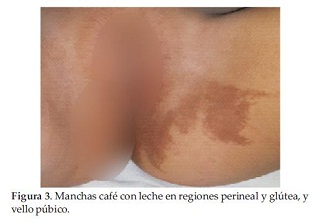
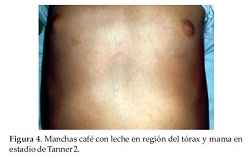
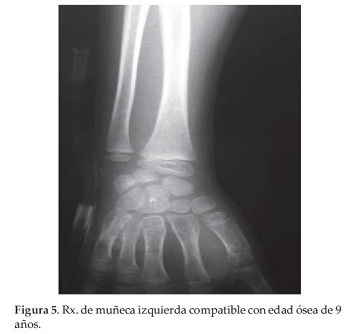
Se constatan manchas café con leche en regiones del tórax anterior, perineal y dorsolumbar, Tanner 2 mamario y púbico (Figura 3 y 4), con antecedente de sangrado vaginal en 2 oportunidades 1 mes antes y con Rx de muñeca izquierda compatible con edad ósea de 9 años (Figura 5).
RMN cerebral: microadenoma hipofisiario (Figura 6). Hormonas: TSH 1.46 (0.3-3.6), LH 0.58 (0.7-155), FSH 0.57 (2.2-100), GH 1.5 (0.06-6.8), Parathormona 45.6 (13.5-39.5). Ante signos de pubertad precoz, manchas café con leche e imágenes (Rx y TAC) compatibles con lesiones fibrosas óseas múltiples, se diagnóstica Síndrome de McCune-Albright, además del microadenoma hipofisiario. La paciente recibe pamidronato, calcio más vitamina D3, y realiza seguimiento con controles periódicos de RMN cerebral por el microadenoma, sin tratamiento hormonal.

DISCUSIÓN
El Síndrome de McCune-Albright (SMA) es una rara entidad que se caracteriza por displasia fibrosa ósea (DFO) poliostótica, lesiones cutáneas hiperpigmentadas y endocrinopatías, la más frecuente es la pubertad precoz y sobre todo en niñas, además de hipertiroidismo, exceso de hormona del crecimiento (GH), pérdida renal de fosfato con o sin raquitismo/osteomalacia y Síndrome de Cushing. En raras ocasiones, otros pueden afectarse (hígado, corazón, paratiroides, páncreas).
Mientras que el SMA es raro, la DFO no lo es, y puede afectar a un solo sitio (DFO monostótica), o varios sitios (DFO poliostótica). Muy rara vez la pubertad precoz se puede asociar a manchas café con leche en la piel en ausencia de DFO (aproximadamente 1% de los casos), pero en general, la DFO parece ser el componente más común del SMA(1-3).
La pubertad precoz es la endocrinopatía más frecuente, es el resultado de una función gonadotropina independiente, autonómica, de ovarios o testículos; más común en el sexo femenino con presencia de ginecomastia y sangrado vaginal. A causa de la exposición excesiva a los estrógenos se incrementa la velocidad de crecimiento y se adelanta la madurez esquelética, lo que trae como consecuencia una menor estatura final. Se han descrito muchos otros trastornos endocrinológicos, caracterizados por hiperfuncionamiento hormonal, tales como hiperplasia adrenal, hipertiroidismo, hiperparatiroidismo, hiperprolactinemia o adenomas hipofisarios secretores de hormona del crecimiento(9,10).
La displasia fibrosa ósea monostótica o poliostótica puede afectar cualquier hueso, pero principalmente el esqueleto axial, a las extremidades, macizo cráneo-facial, tibia y fémur proximal, produciendo dolor crónico, deformidad, asimetría o fracturas espontáneas, los cuales aparecen antes de los 10 años de edad.
La patogénesis de las lesiones óseas no es clara, se cree que la proteína Gsα activada por mutaciones puede ejercer su efecto patogénico sobre los osteoclastos o fibroblastos, imitando los efectos normales de regulación de la hormona paratiroidea, calcitonina u otras hormonas produciendo abundantes células similares a los fibroblastos con mínima matriz extracelular, exceso de células pro-osteogénicas que maduran en osteoblastos anormales lo que provoca un patrón óseo desorganizado, similar a una "sopa alfabética" o de "letras chinas", el osteoide es de forma irregular (retorcido) con un estroma fibroso muy celular(4,11).
Hasta el presente no existe tratamiento contra el problema molecular específico (inapropiada activación de la subunidad Gsa) de esta enfermedad. El tratamiento del SMA requiere un abordaje multidisciplinario de ortopedistas, endocrinólogos, psicólogos y pediatras o internistas según la edad del paciente.
La displasia fibrosa en los pacientes asintomáticos no requieren un tratamiento específico, sólo observación. La mayoría de las fracturas se tratan con tracción, las del extremo proximal del fémur requieren fijación interna. Las grandes deformidades dolorosas se tratan con osteotomías correctoras, aunque el porcentaje de recurrencia es elevado. La displasia fibrosa poliostótica severa con dolores óseos se trata con bifosfonatos. Datos preliminares sugieren que alivian el dolor, reducen la frecuencia de las fracturas patológicas y enlentecen la evolución de la enfermedad ósea(12-14).
Es importante conocer esta asociación de signos a fin de obtener un diagnóstico precoz y manejo adecuado.
REFERENCIAS
1. Albright F, Butler AM, Hampton AO, Smith P. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females. N Engl J Med. 1937;216:727-46. [ Links ]
2. Pitarch Bort G, Laguna Argente C, Martín González B, Febrer Bosch MI, Alegre de Miquel V. Síndrome de McCune-Albright. Med Cutan Iber Lat Am. 2009;37(3):144-46. [ Links ]
3. Marrero Riverón LO, Rondón García V, Melo Víctores M, Chao Carrasco LA, Roché Egües HE, Roche Sánchez JL. Diagnóstico y seguimiento evolutivo de un paciente con Enfermedad de McCuneAlbright. Rev Cubana Ortop Traumatol [Internet]. 2005 [Citado 20 Dic 2013]; 19(2). Disponible en: http://scielo.sld.cu/scielo.php?pid=S0864-215X2005000200011&script=sci_arttext. [ Links ]
4. Dorfman HD, Czerniak B. Fibroosseous Lesions. In: Dorfman HD, Czerniak B, eds. Bone Tumors. St Louis (MO): Mosby; 1998. p. 441-91. [ Links ]
5. Levine MA. Clinical implications of genetic defects in G proteins: oncogenic mutations in G alphas as the molecular basis for the McCune-Albright syndrome. Arch Med Res. 1999;30:522-31. [ Links ]
6. Kumar V, Abba AK, Fausto N. Fibrous and osseous fibrous tumors. In: Kumar V, Fausto N, Abbas A, eds. Robbins and Cotran pathologic basis of disease. 7th ed. Hardcover: Elsevier; 2005. p.1359-61. [ Links ]
7. Weinstein LS, Chen M, Liu J. Gs ( ) mutations and imprinting defects in human disease. Ann NY Acad Sci. 2002;968:173-97. [ Links ]
8. Lee SE, Lee EH, Park H, Sung JY, Lee HW, Kang SY, Seo S, Kim BH, Lee H, Seo AN, Ahn G, Choi YL. The diagnostic utility of the GNAS mutation in patients with fibrous dysplasia: meta-analysis of 168 sporadic cases. Hum Pathol. 2012;43(8):1234-42. [ Links ]
9. Mastorakos G, Mitsiades NS, Doufas AG, Koutras DA. Hyperthyroidism in McCune-Albright syndrome with a review of thyroid abnormalities sixty years after the first report. Thyroid. 1997;7(3):433-39. [ Links ]
10. Akintoye S, Chebli C, Booher S, Feuillan P, Kushner H, Leroith D, Cherman N, Bianco P, Wientroub S, Robey PG, Collins MT. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J Clin Endocrinol Metab. 2002;87(11):5104-12. [ Links ]
11. Hart ES, Kelly MH, Brillante B, Chen CC, Ziran N, Lee JS, Feuillan P, Leet AI, Kushner H, Robey PG, Collins MT. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res. 2007;22:1468-74. [ Links ]
12. Ippolito E, Bray EW, Corsi A, De Maio F, Exner UG, Robey PG, Grill F, Lala R, Massobrio M, Pinggera O, Riminucci M, Snela S, Zambakidis C, Bianco P. Natural history and treatment of fibrous dysplasia of bone: a multicenter clinicopathologic study promoted by the European Pediatric Orthopaedic Society. J Pediatr Orthopaedics. 2003;12:155-77. [ Links ]
13. Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003;88:4569-75. [ Links ]
14. Chan B, Zacharin M. Pamidronate treatment of polyostotic fibrous dysplasia: failure to prevent expansion of dysplastic lesions during childhood. J Pediatr Endocrinol Metab. 2006;19:75-80. [ Links ]

