










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPediatría (Asunción)
On-line version ISSN 1683-9803
Pediatr. (Asunción) vol.41 no.1 Asunción Apr. 2014
CASO CLÍNICO
Síndrome de Allagile. Presentación de un caso
Alagille Syndrome: a Case Report
Lidia María Ortiz-C, Gloria Celeste Samudio-D(1).
1. Servicio de Consulta Externa Pediátrica, Hospital Central del Instituto de Previsión Social. Asunción, Paraguay.
Correspondencia:Dra. Gloria Celeste Samudio. E-mail: gsamudio.samudio@gmail.com
Recibido: 08/07/2013; Aceptado: 05/12/2013.
RESUMEN
El Síndrome de Allagile, conocido también como síndrome de Alagille-Watson y displasia arterio hepática se caracteriza por una colestasis crónica producida por una hipoplasia de las vías biliares intrahepáticas. Se asocia a malformaciones congénitas cardiacas, renales y esqueléticas en pacientes con un fenotipo peculiar. Estas características constituyen los cinco criterios clásicos de diagnóstico. La biopsia hepática ya no es imprescindible para el diagnóstico, siempre y cuando exista ictericia colestasica. El tratamiento es sintomático. El trasplante hepático debe plantearse solo en caso de falla hepática porque la sobrevida no es buena. Se presenta el caso de una niña con diagnóstico de Síndrome de Allagile, que presentó cuatro de los cinco criterios clásicos.
Palabras clave: Síndrome de Allagile, pediatría, criterios diagnósticos.
ABSTRACT
Alagille syndrome, also known as Alagille-Watson syndrome or arteriohepatic dysplasia, is characterized by chronic cholestasis produced by hypoplasia of the intrahepatic bile ducts. It is associated with congenital heart, kidneys, and skeletal malformations in patients with a particular phenotype. These characteristics constitute the five classical diagnostic criteria. Hepatic biopsy is no longer essential for diagnosis when obstructive jaundice is present. Treatment is palliative. Liver transplant should be considered only in case of liver failure, due to unsatisfactory survival rates. We present the case of a female child diagnosed with Alagille syndrome who presented four of the five classic criteria.
Keywords: Alagille syndrome, children, diagnosis, diagnostic techniques and procedures.
INTRODUCCIÓN
El síndrome de Alagille (SA) es una condición que afecta el hígado, el corazón, la columna vertebral, los ojos, la cara, los riñones y los vasos sanguíneos. Es causado por mutaciones en un gen llamado “Jagged 1” en 94% de los pacientes, o “Notch 2” en 1-2% de los casos.
La mayoría de los pacientes con este síndrome tienen enfermedades hepáticas causadas por escasez de los conductos biliares (1). El diagnóstico se establece por la suma de las alteraciones mayores del síndrome, presentes en el niño, además de la escasez de conductos biliares intrahepáticos demostrado por biopsia.
Son marcadores mayores de la enfermedad la colestasis crónica, fascies característica, el soplo cardiaco, embriotoxon posterior y anomalías vertebrales (2-3).
La evolución del síndrome se caracteriza por episodios recurrentes de colestasis, frecuentemente acompañada de infecciones respiratorias. La desnutrición es común. El óbito se debe generalmente a infecciones o complicaciones cardiovasculares. No existe tratamiento específico. El trasplante hepático es una alternativa actual (4-5).
CASO CLÍNICO
Paciente de sexo femenino nacida por cesárea de madre nulípara sin antecedentes patológicos de importancia. G1 P1. EG 40 semanas. Apgar 8-9 Peso de 2960 gramos. Talla 48 cm. Fue dada de alta a las 48 horas sin complicaciones.
Presentó colestasis al mes de vida. Fue sometida a derivación de Kassai a los 2 meses de edad con diagnóstico de atresia de vías biliares. Ante la mala evolución, al año de edad, retorna a la consulta, se replantea el diagnóstico y se encuentran los siguientes datos:
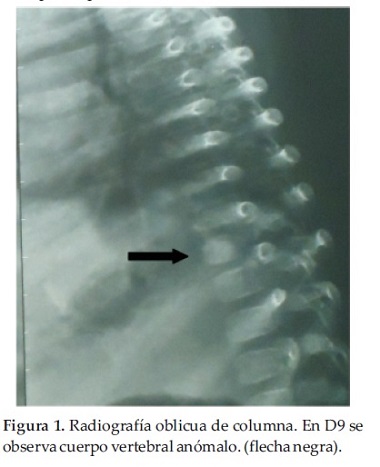
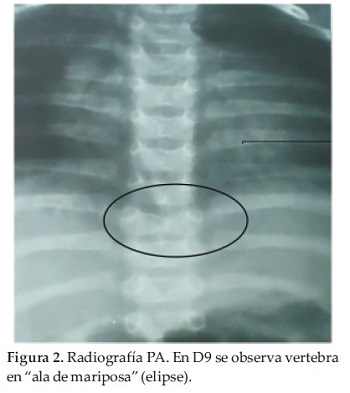
Vertebras en alas de mariposa en las vertebras dorsales 8 y 9 (Figuras 1 y 2).
Se encontró además embriotoxon posterior, con fondo de ojo normal, soplo sistólico en mesocardio con irradiación a la base pero con ecocardiograma normal, fascies característica: nariz y orejas puntiagudas, frente prominente y cara triangulas y mentón fino (simulando cara de duende). Esto, sumado a la colestasis crónica, estableció el diagnóstico. Al año de edad contamos con los siguientes datos laboratoriales: Hb 10 gr/dl, Hto 31 %, GB 3700/mm3, PMNN 31%, L 64%, LR 5%, Plaquetas 342.000/mm3, Glicemia 75 mg/dl, BD 5,7 mg/dl, calcemia 9 mg/dl, fosfatemia 4,73 mg/dl, magnesio 2,8 mg/dl, BT 8,20 mg/dl, fosfatasa alcalina 664 UI/L, GOT 160 UI/L, GPT 103 UI/L. Creatinina 0,43 mg/dl, Urea 20 mg/dl. Tiempo de protrombina 100% y Tiempo de tromboplastina parcial activada 42 segundos. Colesterol 273 mg/dl, Gama GT 106 UI/L, Plasma ictérico.
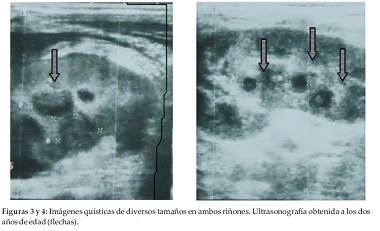
Ecografía abdominal reveló hígado aumentado de tamaño, vías biliares intra y extra hepática no dilatadas, no se observa vesícula biliar. Presencia de formaciones nodulares múltiples en ambos riñones (Figuras 3 y 4) Radiografía de cráneo normal para la edad.
Se solicitó marcadores virales en preparación para trasplante hepático. CMV IgG 250 MEIA, IgM negativo, Toxoplasmosis IgG e IgM negativos, HB anti HB core negativo (MEIA) HB HBS Ag negativo, HA negativo HC negativo, HIV y VDRL negativos. Chagas, EBV Anticuerpo contra capside viral IgG positivo, IgM negativo.
La niña se encontraba ictérica y con prurito intenso cuyo rascado dañaba la piel. El tratamiento recibido fue acido Ursodeoxicolico 75 mg/día junto con suplementos vitamínicos A, C, D y E. A los 2 años y 5 meses de edad presentó un episodio de diarrea aguda, la cual se complicó con un cuadro séptico y falleció por choque irreversible, sin poder realizarse la biopsia hepática.
DISCUSIÓN
La colestasis del lactante es un síndrome clínico caracterizado por ictericia, acolia total o hipocolia, y coluria, que cursa con alteración de la función hepática y elevación de la bilirrubina directa o conjugada (> 2 mg/dl) y de los ácidos biliares séricos (6).
El diagnóstico diferencial de los procesos que causan colestasis en este período de la vida es difícil por la escasa expresividad del hígado y la superposición de los rasgos clínicos, datos analíticos y hallazgos histológicos. La lista de enfermedades que cursan con colestasis es muy extensa e incluye: anomalías estructurales, extrahepáticas e intrahepáticas, que causan obstrucción al flujo biliar, causas infecciosas, tóxicas o metabólicas que alteran los mecanismos de síntesis y excreción de las sales biliares (7).
Uno de los diagnósticos a tener en cuenta es el Síndrome de Allagile, conocido también como síndrome de Alagille-Watson y displasia arterio hepática. Se trata de una colestasis crónica producida por una hipoplasia de las vías biliares intrahepáticas, y se asocia a malformaciones congénitas cardiacas, renales y esqueléticas en pacientes con un fenotipo peculiar. Está considerada una enfermedad genética, con una transmisión autosómica dominante en la que está implicado el cromosoma 20p12.
Los primeros casos fueron descritos en 1973 por Watson y Millar, que notaron la relación existente entre la enfermedad hepática neonatal y la estenosis pulmonar. En 1975 Daniel Alagille, describió el síndrome, identificando las alteraciones de los conductos hepáticos como causa de este padecimiento y describió los rasgos faciales característicos, dados por cara triangular, frente prominente, hipertelorismo, ojos profundos, nariz caída y mentón puntiagudo (1,8,9).
La mayoría de los casos (aproximadamente el 97%) es ocasionada por haploinsuficiencia del gen JAGGED1 (JAG1) sobre el brazo corto del cromosoma 20. En más de la mitad de los casos se trata de mutaciones de novo, inexistentes en los genes de los padres. Un pequeño porcentaje (<1%) es causado por mutaciones en el gen NOTCH2, asociándose a malformaciones renales (10,11).
Las características clínicas son muy variables, sin embargo se han descrito criterios clásicos que permiten sospechar el diagnóstico. Estos criterios, son cinco(12-14): 1) colestasis debida a pobreza de conductos biliares (disminución del número de conductos biliares en relación al número de espacios porta, en la biopsia hepática), 2) enfermedad cardíaca congénita, 3) anormalidades esqueléticas, 4) alteraciones oculares y 5) fenotipo facial característico.
La biopsia hepática ya no es obligatoria si existe colestasis (15). El compromiso hepático se caracteriza por colestasis crónica en alrededor del 95% de los casos, mayoritariamente durante el período neonatal. Existe hepatoesplenomegalia, hiperbilirrubinemia conjugada, hiper colesterolemia, hipertrigliceridemia, y ácidos biliares y aumento de enzimas hepáticas. Suelen presentar retraso del crecimiento, prurito y xantomas. El prurito suele ser invalidante y se presenta a partir del segundo semestre de vida.
Nuestra paciente presentó colestasis marcada desde el nacimiento, lo que derivo en una intervención de Kasai. Al año de vida era portadora de desnutrición severa crónica, que persistió hasta la fecha de deceso. Los datos laboratoriales muestran alteraciones de las enzimas hepáticas, colesterol, hepatomegalia, tal como refiere la literatura. El prurito intenso e inhabilitante fue evidente en nuestro caso, ya que la piel se encontraba muy dañada debido al rascado. La evolución hacia una enfermedad hepática progresiva, cirrosis y fallo hepático, con indicación de trasplante hepático, acontece en el 15% de los casos y se ha reportado casos de carcinoma hepático (16,17).
El compromiso cardíaco está presente en más del 90% de los niños. La cardiopatía congénita más frecuente es la estenosis pulmonar periférica (67%). Nuestra paciente no presentó malformación cardiaca congénita y el soplo que presentaba podría atribuirse a otras causas, una de las cuales podría ser la anemia con la que cursaba (17-19).
El hallazgo ocular más frecuente es el embriotoxón posterior (78-89%), que consiste en una banda prominente, blanca, situada en el ángulo de la cámara anterior del ojo; signo presente en la niña que presentamos. Se identifica con lámpara de hendidura y puede estar presente hasta en el 8-15% de la población normal.
Otras afecciones vinculadas con el SA son: microcórnea, keratocono, distrofia macular congénita, cámara anterior superficial, exotropia, queratopatía en banda y cataratas. La hipopigmentación difusa del fundus retiniano puede ocurrir en hasta un 57% de los pacientes, el moteado del epitelio pigmentario retiniano en el 33% y anomalías de la papila en el 76% (17,20).
Con respecto a las malformaciones esqueléticas, en un 80% de los pacientes se observan las denominadas vértebras en “alas de mariposa” en la radiografía ántero-posterior, ocasionadas por anomalías en la fusión del arco vertebral anterior. Si bien no son patognomónicas del síndrome de Allagile, en esta entidad las “alas de mariposa” son de apariencia simétrica.
Otras anormalidades incluyen adelgazamiento de la distancia interpeduncular de la columna lumbar, espina bífida oculta y fusión de la vértebra adyacente, hemivértebra y ausencia de la duodécima costilla. Se han comunicado craniosinostosis, sinostosis radiocubital, acortamiento de las falanges distales de los dedos que confiere una apariencia fusiforme (13,14,21).
Nuestra paciente poseía vertebras en alas de mariposa de la D8 y D9. Sin evidencia de craneosinostosis. En los rasgos faciales existen características dismórficas leves pero reconocibles, incluidos una frente prominente, ojos hundidos con moderado hipertelorismo, fisuras palpebrales oblicuas ascendentes, puente nasal deprimido, nariz recta con punta bulbosa, orejas grandes, mandíbula prominente y mentón puntiagudo. En algunos niños, la forma de la cara es notablemente triangular, tal como se encontró en nuestra paciente, y fue uno de los motivos que hizo sospechar el diagnóstico (16).
Se han reportado malformaciones vasculares que podrían causar accidentes cerebrovasculares hasta un 15% de los pacientes y han ocasionado el deceso hasta en el 34% de una serie. Las lesiones pueden ser detectadas con una angio resonancia magnética cerebral. Los traumatismos cerebrales menores pueden conducir a hemorragia intracraneana. Otros autores han hecho notar en estos pacientes, el riesgo de sangrado al realizar procedimientos invasivos (22,23).
Nuestra paciente no fue sometida a ningún estudio para demostrar la presencia de malformaciones vasculares, por lo que no podemos saber si estaban presentes o no. Tampoco se refiere antecedente de sangrados.
La afectación renal en estos pacientes puede presentarse como riñones pequeños y ecogénicos, quistes, obstrucción ureteropiélica, nefropatía tubulointersticial y mesangiolipidosis. Los quistes pueden ir desde los simples, sin consecuencias funcionales, los riñones multiquísticos displásicos, hasta los riñones quísticos que conducen a la insuficiencia renal durante la infancia. Se han comunicado compromiso renal vascular y acidosis tubular renal (en hasta el 74% de los casos). En el caso nuestro, la niña presentó riñones poliquísticos diagnosticados por ecografía a los dos años de edad, sin presentar insuficiencia renal ni otro dato que hable de acidosis tubular renal (24,25). Lo llamativo es que estos quistes no se encontraban presentes al año de vida, momento en que cuenta con ecografía normal. Es esto indicativo que los trastornos renales pueden ir apareciendo con el transcurrir del tiempo? Sería un campo interesante a ser investigado.
La insuficiencia pancreática puede presentarse hasta en un 40% de los pacientes. El tratamiento del prurito y xantomas se realiza con ácido ursodexocicolico, con el fin de mejorar calidad de vida, además se debe tener la piel hidratada. El aporte de vitaminas debe ser a grandes dosis. Debido a la malabsorción de ácidos grasos de cadena larga, se debe suplementar las fórmulas con triglicéridos de cadena mediana. El aporte de hidratos de carbono puede mejorar el déficit calórico. Se debe suplementar calcio y zinc (26,27).
En nuestra paciente se cumplió con todos los requerimientos del manejo médico, a pesar de lo cual tanto el prurito como la desnutrición no fueron eliminados.
El tratamiento quirúrgico consiste en derivación externa o trasplante hepático. Este último se plantea en casos de niños con cirrosis e insuficiencia hepática y puede discutirse en los pacientes con colestasis grave, prurito invalidante y pobre calidad de vida (2,4,28).
Nuestra paciente, fue sometida a derivación de Kasai a los 2 meses y si bien se encontraba en programa de trasplante hepático para la fecha del deceso, aun no era candidata al mismo ya que no presentaba insuficiencia hepática.
En conclusión, la niña del caso presentó criterios mayores de SA tales como fascies característica, malformaciones vertebrales, malformaciones renales, colestasis desde el nacimiento y embriotoxon posterior. Si bien no tenemos la biopsia hepática confirmatoria, actualmente este último criterio no es necesario para el diagnóstico. Se agrega al cuadro la desnutrición y trastornos laboratoriales tales como transaminasitis e hipercolesterolemia. Si bien la niña estaba en planes de trasplante hepático, es sabido que el mismo debe plantearse solo en caso de insuficiencia hepática, ya que la sobrevida de estos pacientes, es menor que la verificada para otras indicaciones electivas pediátricas (27,28).
REFERENCIAS
1. Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr. 1975;86:63-71. [ Links ]
2. Kamath BM, Loomes KM, Oakey RJ, Emerick KE, Conversano T, Spinner NB, et-al. Facial features in Alagille syndrome: specific or Alagille syndrome?. Am J Med Genet. 2002;112(2):163-70. [ Links ]
3. Woolf GM, Vierling JM. Disappearing intrahepatic bile duct: the sindrome and their mechanic. Semin Liver Dis. 1993;13:261-75. [ Links ]
4. Kaye AJ, Rand EB, Munoz PS, Spinner NB, Flake AW, Kamath BM. Effect of Kasai procedure on hepatic outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;51:319-21. [ Links ]
5. Arnon R, Annunziato R, Miloh T, Suchy F, Sakworawich A, Sogawa H, et-al. Orthotopic liver transplantation for children with Alagille syndrome. Pediatr Transplantat. 2010;14(5):622-28. [ Links ]
6. Roberts EA. The jaundiced baby. En: Kelly DA, editor. Diseases of the liver and biliary system in children. Oxford: Blacwell-Science; 1999. p. 11-45. [ Links ]
7. Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217-27. [ Links ]
8. Wang JS, Wang XH, Zhu QR, Wang ZL, Hu XQ, Zheng S. Clinical and pathological characteristics of Alagille syndrome in Chinese children. World J Pediatr. 2008;4(4):283-88. [ Links ]
9. Méndez MV, Burastero M, Cuadro MN, Casamayou L, Gutiérrez C, Montano A, et-al. Colestasis neonatal: evolución de los pacientes diagnosticados entre 1983 y 2000 en dos servicios gastroenterológicos de referencia de Montevideo. Arch Pediatr Urug. 2005;76(3):210-21. [ Links ]
10. Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et-al. Alagille syndrome is caused by mutations in human JAGGED1, which encodes a ligand for NOTCH1. Nat Genet. 1997;16(3):243-51. [ Links ]
11. Crosnier C, Driancourt C, Reynaud N, Dhorne-Pollet S, Pollet N, Bernard O, et-al. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology. 1999;116(5):1141-48. [ Links ]
12. Hingorani M, Nischal KK, Davies A, Bentley C, Vivian A, Baker AJ, et-al. Ocular abnormalities in Alagille syndrome. Ophthalmology. 1999;106:330-37. [ Links ]
13. Sanderson E, Newman V, Haigh SF, Baker A, Sidhu PS. Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Pediatr Radiol. 2002;32(2):114-19. [ Links ]
14. Bales CB, Kamath BM, Munoz PS, Nguyen A, Piccoli DA, Spinner NB, et-al. Pathologic lower extremity fractures in children with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;51(1):66-70. [ Links ]
15. Ciocca M, Alvarez F. Síndrome de Alagille. Arch Argent Pediatr. 2012;110(6):509-15. [ Links ]
16. Emerick KM, Rand EB, Goldmuntz E, Krantz ID,Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822-29. [ Links ]
17. Tumpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251-57. [ Links ]
18. McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, et-al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106(20):2567-74. [ Links ]
19. Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, et-al. Familial Tetralogy of Fallot caused by mutation in the JAGGED 1 gene. Hum Mol Genet. 2001;10(2):163-69. [ Links ]
20. Crosnier C, Lykavieris P, Meunier-Rotival M, Hadchouel M. Alagille syndrome: the widening spectrum of arterio hepatic dysplasia. Clin Liver Dis. 2000;4(4):765-78. [ Links ]
21. Olsen IE, Ittenbach RF, Rovner AJ, Leonard MB, Mulberg AE, Stallings VA, et-al. Deficits in size-adjusted bone mass in children with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;40(1):76-82. [ Links ]
22. Emerick KM, Krantz ID, Kamath BM, Darling C, Burrowes DM, Spinner NB, et-al. Intracranial vascular abnormalities in patients with Ala gille syndrome. J Pediatr Gastroenterol Nutr. 2005;41(1):99-107. [ Links ]
23. Lykavieris P, Crosnier C, Trichet C, Meunier-Rotival M, Hadchouel M. Bleeding tendency in children with Alagille syndrome. Pediatrics. 2003;111(1):167-70. [ Links ]
24. Shrivastava R, Williams A, Mikhail A, Roberts D, Richards M, Aithal V. An unusual cause of hypertension and renal failure: a case series of a family with Alagille syndrome. Nephron Dial Transplant. 2010;25(5):1501-06. [ Links ]
25. Davis J, Griffiths R, Larkin K, Rozansky D, Troxell M. Glomerular basement membrane lipidosis in Alagille syndrome. Pediatr Nephrol. 2010;25(6):1181-84. [ Links ]
26. Mattei P, von Allmen D, Piccoli D, Rand E. Relief of in tractable pruritus in Alagille syndrome by partial external biliary diversion. J Pediatr Surg. 2006;41:104-07. [ Links ]
27. Kamath BM, Loomes KM, Piccoli DA. Medical manage ment of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50:580-86. [ Links ]
28. Kamath BM, Schwarz KB, Hadzic N. Alagille syndrome and liver transplantation. J Pediatr Gastroenterol Nutr. 2010;50:11-15. [ Links ]

