










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPediatría (Asunción)
On-line version ISSN 1683-9803
Pediatr. (Asunción) vol.41 no.1 Asunción Apr. 2014
ARTÍCULO ORIGINAL
Lipofuscinosis ceroidea neuronal infantil tardía (Jansky- Bielchowsky). Estudio de casos
Late-Infantile Neuronal Ceroid Lipofuscinoses (Jansky-Bielschowsky Disease): a Study of a Series of Cases
Jorge Ortiz, María Lezcano, Alicia Aldana, Mariana Paredes, Zoilo Morel, Ida Esquivel, Karen Romero, Laura Fretes (1)
1. Cátedra y Servicio de Pediatría. Facultad de Ciencias Médicas. Universidad Nacional de Asunción. San Lorenzo, Paraguay.
Correspondencia: Dr. Jorge Daniel Ortiz Rolón. E-mail: jdor_py@hotmail.com
Recibido: 20/12/2013; Aceptado: 17/02/2014
RESUMEN
Introducción: La lipofuscinosis ceroidea neuronal constituye el grupo de desordenes genéticos neurodegenerativos de depósito más común en la infancia, afecta a niños, adultos jóvenes y tiene herencia autosómica recesiva. Objetivo: Presentar un estudio de casos clínicos de 8 pacientes con lipofuscinosis ceroidea neuronal infantil tardía. Metodología: Estudio de casos clínicos de pacientes cuyos diagnósticos de lipofuscinosis ceroidea neuronal infantil tardía es propuesto a través del cuadro clínico: semiología de las crisis epilépticas, cronología de la aparición de las manifestaciones visuales, motoras, cognitivas y regresión en el neurodesarrollo. Resultados: Caso 1. Niño de 6 años de edad, con pérdida progresiva de la visión desde los 3 años llegando a la amaurosis, epilepsia desde los 4 años, ataxia y dificultades motoras y cognitivas a los 5 años. Electroencefalograma interictal patrón epiléptico. Resonancia Magnética de cráneo con atrofia de cerebro y cerebelo e indemnidad de ganglios basales. Casos 2, 3, 4 y 5 muestran el carácter hereditario, presentándose en 2 miembros de la misma familia. Caso 6, de inicio tardío, fue clasificada posteriormente como tipo 3, forma Juvenil, por análisis mutacional. Caso 7, convulsiones como síntoma inicial a los 3 años, resaltando las dificultades conductuales en el relacionamiento con sus pares lo que motiva la consulta con especialista. Caso 8, con una evolución rápida hacia el deterioro global. Conclusiones: La LCN es una entidad a tener en cuenta ante un paciente entre los 2 y 4 años que presente epilepsia refractaria, regresión del desarrollo y compromiso visual. La presencia de epilepsia mioclónica orienta la búsqueda de la etiología, modifica la conducta terapéutica. No existe aún un tratamiento definitivo para esta enfermedad, el manejo se dirige desde las fases iniciales a mejorar la calidad de vida del paciente y la de su familia con medidas generales y de sostén.
Palabras clave: Lipofuscinosis ceroidea neuronal infantil tardía, epilepsia mioclónica, enfermedades de depósito lisosomal.
ABSTRACT
Introduction: Neuronal ceroid lipofuscinoses is a group of neurodegenerative autosomal recessive genetic disorders caused by storage of lipopigments that is most common in infancy, but that affects children and young adults. Objective: To present a series of 8 clinical cases of patients with late-infantile neuronal ceroid lipofuscinosis. Methodology: We present the clinical cases of 8 patients, whose diagnosis of late-infantile neuronal ceroid lipofuscinosis was made clinically by causation of epileptic crises, chronology of the appearance of visual, motor, and cognitive manifestations, as well as neurodevelopmental regression. Results: Case 1: a 6 year-old boy with progressive loss of vision from age 3 progressing to amaurosis, epilepsy from age 4 years, ataxia and motor and cognitive problems detected at age 5 years, interictal electroencephalogram showing epileptic pattern, and brain MRI showing brain and cerebellum atrophy with basal ganglia unaffected. Cases 2, 3, 4, and 5 show hereditary characteristics, appearing in 2 members of the same family. Case 6, of late-onset, was later classified as type 3, a childhood type, based on analysis of mutations. Case 7: the initial symptom was seizures at age 3 years, with behavior difficulties relating to peers being the complaint that led to consultation with a specialist. Case 8: rapid progression toward overall deterioration. Conclusions: Neuronal ceroid lipofuscinosis should be considered in cases of patients aged 2-4 years who present refractory epilepsy, developmental regression, and affected vision. The presence of myoclonic epilepsy guides the search for a cause and modifies the treatment management. No definitive treatment yet exists for the disease, and management is initially directed at improving quality of life for the patient and his or her family using general and supportive measures.
Keywords: Neuronal ceroid lipofuscinosis, late-infantile neuronal ceroid lipofuscinosis, myoclonic epilepsy, lysosomal storage disease.
INTRODUCCIÓN
En el cotidiano andar del pediatra la evaluación del neurodesarrollo debe constituir un momento central de la consulta. Estamos más habituados a ver desfasajes en las adquisiciones de los hitos del desarrollo, sin embargo, cuando lo que acontece es la pérdida de habilidades ya adquiridas y, desde nuestro lugar, nos toca asistir el deterioro progresivo llegando incluso a la muerte prematura urge el deseo de poder asistir en mejor modo. Una de las formas de lograr esto es conociendo ciertas patologías que tomadas en conjunto distan mucho de ser raras, la agresividad de sus manifestaciones movilizan un deseo de búsqueda del diagnóstico, pronóstico y de ser posible un tratamiento ya sea curativo o paliativo.
Las lipofuscinosis fueron agrupadas inicialmente bajo el nombre de “idiocia familiar amaurótica” intentando destacar así sus dos principales características: compromiso intelectual y visual. La primera sistematización y correlación anatomoclínica la hizo el médico francés Frederick Batten en 1903, por lo que el cuadro llevó inicialmente su nombre. En el año 1969, Zeman y Dyken acuñaron el término “lipofuscinosis ceroidea neuronal” (LCN) y luego de múltiples descripciones de otras patologías que compartían características similares, se propuso la primera clasificación realizada por Norman y Wood. Con posterioridad se han planteado otras con el aporte de la genética y de la morfología microscópica (1,2).
La característica común de estas devastadoras condiciones es la acumulación lisosomal de un lipopigmento autofluorescente, la lipofuscina (3) pese a que no es la causa del daño celular.
Constituyen un grupo de desordenes genéticos neurodegenerativos con herencia autosómica recesiva que afecta a los niños y a los adultos jóvenes. La forma autosómica dominante es una variante en adultos (4).
Se caracterizan por deterioro global mental y motor, pérdida de la visión y epilepsia que desencadenan en la muerte (5). Conjuntamente son el desorden neurodegenerativo de depósito más común en la infancia, posee una prevalencia de hasta 1 en 12500 personas en algunas poblaciones (6,7).
Las LCN son enfermedades que presentan amplia variabilidad clínica y se caracterizan por ser genéticamente heterogéneas. Las mutaciones en un mismo gen originan fenotipos diferentes: mutaciones comunes se asocian a un fenotipo clásico, mientras que mutaciones menos frecuentes pueden originar un fenotipo variante. Además, existen mutaciones asociadas a familias de un área geográfica determinada (2).
En las últimas dos décadas, los importantes avances en la investigación genético-molecular han logrado la identificación de varios genes responsables de las diferentes formas clínicas en la edad pediátrica (CLN10/CTSD, CLN1/PPT1, CLN2/TPP1, CLN3, CLN5, CLN6, CLN7/MFSD8, CLN8 entre otros) y actualmente el análisis mutacional proporciona el diagnóstico definitivo de cada una de estas entidades y permite establecer correlaciones genotipo-fenotipo (8).
En función de la edad de comienzo, el curso clínico y la morfología ultraestructural, las LCN se han clasificado en cuatro tipos principales:
1. LCN infantil (enfermedad de Haltia-Santavuori, LCN1), hallada predominantemente en Finlandia, se inicia entre los 6 meses y los 2 años generalmente, se relaciona con diversas mutaciones en el gen LCN1, en el cromosoma 1p32, que determinan el déficit de una enzima lisosomal denominada tioesterasa proteinopalmitoil 1 (o PPT1).
2. LCN infantil tardía (LCNIT) (enfermedad de Jansky-Bielschowsky, LCN2), se inicia entre los 2 y 4 años de vida con crisis tónico-clónicas generalizadas, ausencias o crisis parciales secundariamente generalizadas, aunque su característica típica es la mioclonía que aparece posteriormente junto a la pérdida cognitiva, ataxia, signos extrapiramidales y piramidales, deterioro rápidamente progresivo de la visión por atrofia óptica. Las crisis convulsivas se asocian a pérdida o falta de adquisición de habilidades del desarrollo psicomotor. El paciente puede fallecer en este primer episodio o derivar a estado vegetativo permanente por años.
3. LCN juvenil (enfermedad de Batten, enfermedad de Spielmeyer-Vogt-Sjorgenkjnng, LCN3) es la más común de las lipofuscinosis en la población norteamericana, correspondiendo al 45 a 52% de los casos. En la forma clásica los primeros síntomas aparecen entre los 4 y 7 años de vida, no existe aún una alteración enzimática identificada pero se sabe que el gen LCN3 se encuentra en el cromosoma 16p12 y codifica para una proteína estructural del lisosoma.
4. LCN del adulto (enfermedad de Kufs) los síntomas iniciales aparecen alrededor de los 30 años, siendo una de sus características relevantes la indemnidad ocular. Clásicamente existen 2 fenotipos: un cuadro caracterizado por epilepsia mioclónica progresiva con demencia, ataxia y signos piramidales y extrapiramidales tardíos, y otro con trastornos del comportamiento y demencia que se pueden asociar a ataxia y signos extrapiramidales (1).
Se han descrito además variantes o formas atípicas que representan alrededor de un 20% de las LCN en diferentes poblaciones, generalmente distribuidas entre los grupos infantil tardío y juvenil. Para la forma infantil tardía se han descrito la variante finlandesa (LCN5) y la variante LCN6. Existe otra variante conocida como LCN8, epilepsia del norte o epilepsia progresiva con retardo mental (EPRM) (1).
Las mutaciones en el gen CLN2 en el cromosoma 11p15, conduce al cuadro clásico de Lipofuscinosis ceroidea neuronal infantil tardía, también denominada enfermedad de Jansky-Bielschowsky (7), estas mutaciones determinan la deficiencia de una enzima lisosomal, la tripeptidil-peptidasa o TPP1 (1), componente de una familia de serina-carboxilproteinasas cuya función es eliminar el aminoácido terminal de las proteínas sometidas a degradación lisosomal favoreciendo el acumulo de la subunidad c de ATP sintetasa mitocondrial (8,9). Los cambios producidos a nivel celular y molecular dan cuenta de una acumulación de pigmento a nivel celular y disfunción mitocondrial que se relacionan con estrés oxidativo que induce la fragmentación mitocondrial en células deficientes de TTP 1 (10). Se han realizado estudios que demuestran que existe una concentración elevada en forma significativa de superóxido y de peróxido de hidrogeno en los fibroblastos con mutaciones CLN2 en comparación con fibroblastos sin tal mutación, esto causa además disfunción de peroxisomas con un descenso de la actividad de catalasa lo cual contribuye al daño y a la degeneración celular (11).
La presente casuística tiene por objetivo presentar un estudio de casos clínicos de 8 pacientes con lipofuscinosis ceroidea neuronal infantil tardía atendidos en centros de referencias de nuestro país.
METODOLOGÍA
Estudio de casos clínicos de 8 pacientes atendidos en centros de referencias de nuestro país en un periodo de 15 años (1998 a 2013); cuyos diagnóstico de lipofuscinosis ceroidea neuronal infantil tardía es propuesto a través del cuadro clínico: semiología de las crisis epilépticas, cronología de la aparición de las manifestaciones visuales, motoras, cognitivas y regresión en el neurodesarrollo.
RESULTADOS
En un periodo de 15 años (1998 a 2013) han sido evaluados 8 pacientes, con criterios clínicos, radiológicos y electroencefalográficos de LCN que fueron analizados a raíz del caso 1 por ser quien motivó esta presentación (desde ahora denominado caso índice).
Caso N° 1 (CASO ÍNDICE)
Escolar de 6 años de edad, sexo masculino, procedente de Luque, hijo de padres no consanguíneos, acudió por primera vez a nuestro servicio en junio de 2013 por historia de convulsiones tres días antes del ingreso, en 3 oportunidades. Las mismas se repiten dos horas antes, tipo clónica generalizada de 20 minutos de duración en vigilia. Ingresó en brazos del padre convulsionando.
Una vez estabilizado el paciente se lo observaba hipoactivo, no conectado, sin respuesta verbal ni contacto visual, con respuesta inespecífica al dolor y exacerbación de las crisis convulsivas ante estímulos auditivos.
Presentaba nistagmo horizontal. Al fondo de ojo se constató atrofia severa del nervio óptico. En lo motor hipertonía distal de miembros inferiores, pie equino fijo, reflejos de estiramiento muscular aumentados, signo de Babinski bilateral presente, marcha ausente, se observaban movimientos involuntarios, breves, como sacudidas en miembros, dificultad para deglutir, hipotrofia generalizada con índice de masa corporal por debajo del percentil adecuado para la edad.
El paciente acudió con el diagnóstico previo de Neuropatía Óptica de Leber formulado en mayo de 2012 en otro centro hospitalario en relación a la amaurosis adquirida y la epilepsia secundaria.
En el interrogatorio del desarrollo psicomotor del niño se evidenciaban adquisiciones de hitos adecuados en la primera infancia, tuvo sostén cefálico y sonrisa social a los 3 meses, se sentó a los 5 meses, pronunció sus primeras palabras a los 8 meses, se paró a los 11 meses y caminó al año de vida, la única limitación que presentaba era en el área del lenguaje ya que a los tres años solamente decía frases sueltas.
Se recabaron datos patológicos de valor en el niño consistentes en pérdida progresiva de la visión desde los tres años de edad. El primer episodio convulsivo se produjo en el 2012, a la edad de 4 años, el mismo fue interpretado como una convulsión febril, el siguiente episodio se presentó 6 meses antes del ingreso y fue tratado con ácido valproico y clonazepam. Posteriormente se agregó al cuadro ataxia progresiva y dificultades en la marcha a los 5 años de vida con deterioro progresivo de las habilidades cognitivas.
Se indagaron sobre antecedentes familiares no obteniéndose datos de epilepsia y/o ceguera en los miembros de la familia.
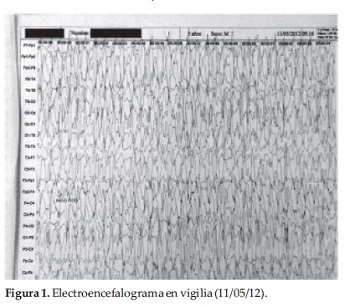
Acudió con Electroencefalograma realizado en vigilia (Figura 1), en el mismo se evidencia importante actividad comicial interictal en forma de Espigas, complejos Punta Onda, Polipuntas de alto voltaje durante todo el trazado, comprometiendo la organización de fondo, no se constata ritmo Alpha posterior, sin gradiente anteroposterior, no se realiza estimulación fótica a baja frecuencia.
Poseía además un resultado de Potencial Visual Evocado realizado 3 años antes de la internación en nuestro servicio que informa potenciales no reproducibles, desorganizado, configuración anormal. No puede determinarse latencia ni amplitud (Figura 2).

En nuestro servicio se plantearon los diagnósticos al ingreso de Epilepsia Sintomática de etiología a determinar, Neuropatía óptica de Leber y Desnutrición calórica proteica severa.
Durante su internación volvieron a repetirse los episodios convulsivos, siendo éstas de difícil manejo quedando con triple terapia anticonvulsivante: ácido valproico, carbamazepina y difenilhidantoína.
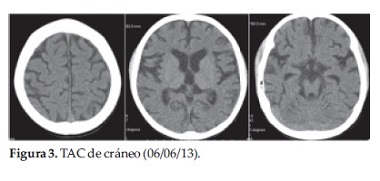
En el primer día de internación se realizó una Tomografía de cráneo en la que se observaba disminución volumétrica del parénquima cerebral, surcos profundos. Cerebelo con disminución volumétrica del parénquima y evidencia de folias cerebelosas. Hallazgos compatibles con atrofia de cerebro y cerebelo (Figura 3).
Se descartó la Neuropatía Óptica de Leber ante ausencia de elementos clínicos y antecedentes familiares compatibles.
Al segundo día de internación se replantearon los diagnósticos, desde una perspectiva clínica nos encontrábamos ante un niño con una patología degenerativa global que afectaba tanto a las habilidades cognitivas, motoras y visuales, cuyo motivo de ingreso fue la epilepsia refractaria; quedando así con los siguientes diagnósticos: 1) Síndrome piramidal, 2) Amaurosis adquirida bilateral, 3) Demencia, 4) Epilepsia sintomática.
Se reinterrogó a los familiares y se obtuvieron datos compatibles con Epilepsia Mioclónica Progresiva por lo que se suspendió la administración de difenilhidantoina y de carbamazepina adicionándose topiramato con buena respuesta. Durante la internación se constataron varios episodios convulsivos mioclónicos intensos y amplios a predominio de miembros.
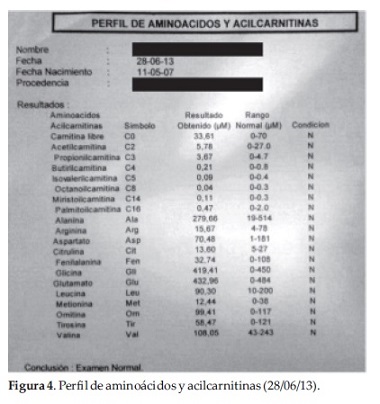
Se investigaron otras posibles etiologías que puedan integrar la sintomatología del paciente (pérdida de la agudeza visual hasta llegar a la amaurosis, pérdida de las habilidades motoras, síndrome piramidal y epilepsia refractaria), se solicitó screening metabólico con Perfil de aminoácidos y Acil Carnitina con resultados en rango (Figura 4).
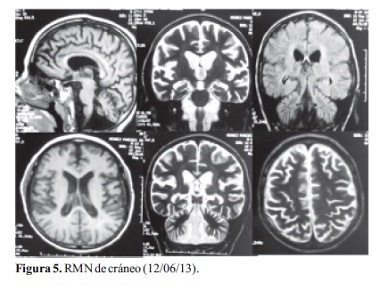
Al séptimo día se le realizó una Resonancia Magnética de cráneo (Figura 5) evidenciándose importantes modificaciones atróficas de cerebro y muy marcadas de cerebelo, no correlativas con la franja etaria. Indemnidad de Ganglios Basales.
Con los datos clínicos: deterioro neurológico, presencia de epilepsia mioclónica progresiva, compromiso visual, motor y cognitivo, complementados con estudios de neuroimagen que mostraban atrofia supratentorial y llamativamente de cerebelo, potencial evocado visual y electroencefalograma patológicos se planteó el diagnóstico presuntivo de Lipofuscinosis Ceroidea Neuronal, compatible con el tipo infantil tardío (enfermedad de Jansky-Bielschowsky) por la edad del paciente y por la indemnidad de los Ganglios Basales en la Resonancia Magnética.
Permaneció internado por 13 días y fue dado de alta con mejoría de las convulsiones, medicado con topiramato y valproato, en planes de seguimiento por Neurología, Pediatría y Fisioterapia.
Caso N° 2
A. A. Paciente de sexo masculino, producto de la gesta 4, previamente sano, sin antecedentes familiares de valor, presenta como primera sintomatología crisis convulsivas, la primera de ellas con fiebre a los 2 años y 3 meses de edad, de tipo tónica generalizada. Posteriormente fue presentando convulsiones afebriles. Primera evaluación oftalmológica sin hallazgos patológicos.
A los 3 años 11 meses el deterioro abarca: temblor, dificultad en la marcha, ataxia progresiva. Deja de caminar a los 4 años de edad, tiempo en el cual ya presentó atrofia óptica y compromiso cognitivo.
Electroencefalograma patológico interictal, con desorganización del ritmo de fondo, actividad comicial focal y generalizada de espigas, Complejos punta onda lenta. No se realiza Fotoestimulación a baja frecuencia. La Resonancia Magnética de Cráneo evidenciaba atrofia cerebral y de cerebelo, gracias al cuadro clínico se condujo a la presunción clínica.
El tiempo transcurrido entre el primer síntoma y la sospecha diagnóstica fue de 2 años. En el transcurrir de su enfermedad presentó diferentes tipos de crisis: atónicas y mioclónicas que no habían sido registradas en su historial hasta el momento en que se hicieron muy frecuentes y se volvieron resistentes a los fármacos utilizados: valproato, benzodiacepina, etc. Presentó síndrome Pseudobulbar, con marcada dificultad para la deglución que derivó en la realización de gastrostomía.
Como dato particular presentaba priapismo. Falleció a la edad de 12 años 8 meses por un cuadro de Neumonía en nuestro hospital. Cursaba él con 7 años de edad cuando se manifiesta la misma entidad en su hermana menor.
Caso N° 3
D. A. Paciente de sexo femenino, hermana del paciente antes referido. Producto de la gesta 5 (familia compuesta por 2 hijas mujeres 3 hijos varones). Desarrollo normal en la primera infancia, logra caminar a los 10 meses, primeras palabras antes del año.
La madre supo el diagnóstico del hijo mayor y de la posibilidad de recurrencia cuando esta niña tenía 1 año de edad. A los 3 años y 1 mes de vida inician las convulsiones. Crisis epilépticas de tipo parcial compleja con componente motor. Se repiten al mes, los eventos aumentan en duración llegando a un promedio de 20 minutos. Al llegar a los 6 meses del inicio del cuadro aparecen crisis atónicas muy frecuentes. Fue medicada con Valproato y Benzodiacepina a los que se agrega Topiramato con mejoría en el control de las crisis. La instalación de nuevos tipos de crisis coincide con el inicio de la ataxia troncal, temblor de miembros superiores, mirada poco expresiva, inicia dificultad en la deglución.
La Resonancia Magnética de cráneo indicaba atrofia cerebral y cerebelosa. Fue evaluada en un centro de referencia en Sudamérica (Santiago de Chile) donde le realizan biopsia de piel con hallazgo ultraestructural compatible con LCN 2, así mismo EEG que reporta actividad epiléptica interictal de punta onda lenta aislada o en series de localización generalizada con mayor voltaje en regiones anteriores y multifocal independiente: temporal izquierda, occipital derecha y frontal izquierda.
Deja de caminar a los 6 años. Reportan crisis mioclónicas a los 6 años 10 meses. Con gastrostomía ya en ese momento. Mantiene la capacidad de seguir con la mirada y visión luz. A los 8 años ya muy deteriorada, descontrol de crisis: parciales, atónicas, mioclónicas, tónicas generalizadas. Fondo de Ojo con atrofia óptica. En este punto fallece su hermano.
Evoluciona hacia mioclonías constantes. Se agrega insomnio y crisis de llanto que sugieren dolor en diferentes horarios. Se investigan posibles etiologías infecciosas, reflujo gastroesofagico y posturales, sin hallazgo positivo.
Se sugiere internación en clínica de cuidados paliativos a la edad de 11 años para adecuado manejo y contención familiar pero no se realiza.
Caso N° 4
N. C. paciente sexo femenino, previamente sana. Inicia el cuadro con crisis convulsivas a los 3 años 4 meses, de tipo parcial motora y generalizada.
A los 6 años se tiene la sospecha diagnóstica, 3 años después del inicio de los síntomas, por la presencia de multicrisis, deterioro progresivo motor, cognitivo y visual. Se detecta atrofia óptica a los 6 años. EEG con actividad epiléptica interictal focal y generalizada. Resonancia Magnética de cráneo con atrofia cerebral y cerebelosa. No afectación de ganglios basales.
Deterioro franco desde los 8 años con afectación cognitiva, dificultad para la deglución que lleva a gastrostomía, crisis mioclónicas refractarias. Fallece a los 12 años por complicaciones respiratorias. La hermana menor ya cursaba con síntomas iniciales de esta patología al momento de su fallecimiento.
Caso N° 5
N. C. Paciente de sexo femenino, hermana de la niña correspondiente al caso 4.
Inicia con crisis convulsivas tónicas generalizadas a los 3 años de edad, momento en el que se plantea el diagnóstico. Las crisis mioclónicas fueron detectadas a los 4 años de vida, así como las atónicas.
Electroencefalograma patológico patrón comicial focal y generalizado. Resonancia Magnética de cráneo con atrofia de cerebro y cerebelo. Atrofia óptica detectada a los 5 años. Recibió cuidados en una clínica de cuidados paliativos donde estuvo hasta su fallecimiento.
Es de destacar que el hecho de tener en un solo lugar apoyo nutricional, pediátrico, neurológico, fisioterapia motora y respiratoria y por sobre todo contención humana marcó una gran diferencia en la manera de afrontar la muerte en esta madre y en toda la familia.
Caso N° 6
M. A. Paciente de sexo femenino.
Niña sana hasta los 4 años de edad, siguió yendo al colegio hasta los 5 años. Inició con crisis convulsivas de tipo parcial motora con versión cefálica a izquierda de 20 minutos. Durante los 2 primeros años las crisis se presentaban 1 vez al mes, siempre en el hemicuerpo izquierdo. Cuando estas se hicieron más frecuentes la madre relata la dificultad visual y el compromiso cognitivo. Se agregan crisis atónicas en vigilia y las mioclónicas sólo en sueño.
A los 6 años se plantea Epilepsia refractaria, Encefalopatía Progresiva, patología neurodegenerativa a descartar. Tiempo transcurrido entre el inicio de los síntomas y sospecha diagnóstica fue de 2 años.
A los 7 años acuden a un centro de referencia en Sudamérica (Buenos Aires), le solicitan Biopsia de piel que no se realiza. El EEG reportaba paroxismo de Polipuntas, Punta Onda de alta frecuencia, atenuación paroxística de voltaje. Estimulación Fótica negativa. Posteriormente se realiza análisis mutacional con delección CLN3-1.02 kb con la que se confirma el diagnóstico de Lipofuscionsis ceroidea neuronal tipo III. La progresión fue hacia un estado de dependencia, convulsiones no controladas y realización de gastrostomía.
Caso N° 7
M. Z. Paciente de sexo femenino. Como antecedente familiar de valor, la madre se encontraba con afectación de las funciones cognitivas y demencia. Sin datos de convulsiones o síntomas que puedan corresponder a un síndrome extrapiramidal.
La niña inicia con cuadro convulsivo a los 3 años de edad. Crisis con versión cefálica secundariamente tónica generalizada. Cursó el jardín a los 4 años con dificultades para la adaptación grupal. A los 5 años la derivan a una escuela para niños con necesidades especiales donde reportan trastornos en la motricidad fina y déficit de atención.
En la primera evaluación de un centro de referencia en nuestro país la describen como una niña inquieta, por momentos con reacciones agresivas hacia los demás, actitud indiferente al medio, temblor de manos, ataxia, dismetría, marcha con dificultad.
Sus primeros diagnósticos fueron Déficit de Atención, Crisis Convulsiva, Síndrome Cerebeloso, Retraso Psicomotor. En el siguiente control se replantean diagnósticos hacia una Encefalopatía Progresiva. La resonancia magnética es informada como normal pero con aumento de surcos corticales.
La terapista ocupacional describe mirada perdida, por momentos sigue algunas consignas. Se presenta el caso a neurólogo infantil quien describe atrofia cerebral y cerebelosa importante, secuencia en la aparición de los síntomas: niña sana, posteriormente convulsiones, compromiso cognitivo-conductual, deterioro motor, visual.
Se plantea Lipofuscinosis ceroidea como sospecha diagnóstica 2 años después de la primera sintomatología.
Al año siguiente el deterioro fue importante ya con falta de respuesta a los estímulos, dificultad para la deglución. Presentó varias internaciones por Estado convulsivo y complicaciones respiratorias. Con el transcurrir de la enfermedad se presentaron crisis de llanto, irritabilidad por momentos. Mioclonías frecuentes y bruscas. Actualmente con 10 años de vida.
Caso N° 8
S. G. Paciente de sexo femenino.
Inicia con crisis convulsivas a los 2 años 4 meses. Presentó varios tipos de convulsiones de manera precoz, atónicas, parciales motoras, ausencias atípicas. Progresa a ataxia y dificultades cognitivas que llevan al planteamiento de Encefalopatía Epiléptica, Sindrome de Lenox Gastaut. EEG anormal interictal, actividad comicial generalizada predominante con Espigas y complejos Punta Onda lenta. Sin respuesta a la medicación, valproato, topiramato y clobazan.
Presentó dificultades para la deglución, desnutrición a los 3 años de edad. Resonancia Magnetica de cráneo con atrofia cerebral y cerebelosa.
Fue evaluada en un centro de referencia de Sudamérica (Buenos Aires) donde se plantea Encefalopatía progresiva heredodegenerativa por la presencia de diferentes tipos de crisis: atónicas, mioclónicas, parcial motora, compromiso visual y motor se orienta hacia Epilepsia mioclónica progresiva, dentro de ellas a LCN. Actualmente se encuentra sin seguimiento.
DISCUSIÓN
En un periodo de 15 años (1998 a 2013) han sido evaluados 8 pacientes, con criterios clínicos, radiológicos y electroencefalográficos de LCN que fueron analizados a raíz del caso 1, denominado caso índice.
El promedio de edad del inicio de la sintomatología se sitúa en los 3,1 años, coincidente con el inicio de los síntomas en una serie española de 11 pacientes (12) que situaba la edad entre los 18 meses y los 3,7 años. El debut de las convulsiones se dio con fiebre al inicio del cuadro en todos los pacientes de la citada investigación al igual que en los casos 1 y 2 presentados en este trabajo, sin embargo dicha eventualidad no es frecuente (13). El sexo predominante fue el femenino (87%), lo cual no es reportado en la literatura. En nuestra serie el promedio de tiempo entre el primer síntoma y la sospecha clínica de LCN fue de 2 años.
Hemos encontrado recurrencia en 2 familias (casos 2, 3 y 4, 5) y en el caso 7 la madre de la niña posee el diagnóstico de demencia sin poder especificar más datos sobre la misma (3).
El caso índice acudió a nuestro servicio con el diagnóstico de Neuropatía Óptica Hereditaria de Leber (NOHL), basado en la amaurosis adquirida y en el informe de los potenciales evocados visuales (Figura 1). Al analizar la bibliografía disponible (11) se descarta dicha etiología por varias razones, en primer lugar porque generalmente se produce en personas sanas, no siendo este el caso de nuestro paciente aunque una pequeña proporción puede presentar problemas neurológicos menores como reflejos de estiramiento muscular alterados y mioclonías (14), además no se obtienen datos familiares de ceguera o pérdida de la visión, siendo esta una entidad de herencia materna producida por mutaciones puntuales en el cromosoma mitocondrial de genotipo homoplásmico y de baja penetrancia. En la NOHL se describe una microangiopatía peripapilar telangiectásica, con atrofia de fibras nerviosas, con alteración del campo visual y pérdida posterior de la visión (15) dichas características no fueron halladas en nuestro paciente.
En la LCN el examen oftalmológico pone de manifiesto degeneración macular, acúmulos de pigmentos en la retina periférica, estrechez del árbol vascular y atrofia óptica (7,8) lo cual se observa en la totalidad de los pacientes analizados en nuestro estudio.
Debido a la edad de inicio se clasificó a nuestro caso índice como subtipo Infantil Tardío que es caracterizado según Williams (13) desde el comienzo por convulsiones y demencia progresiva a la edad de 2 a 4 años. La ataxia y las mioclonías son hallazgos constantes. Los niños quedan incapacitados a la edad de 3 a 6 años, en dicho momento las anormalidades oftalmológicas suelen observarse. Degeneraciones maculares y retinianas asociadas a atrofia óptica ocurren causando ceguera a la edad de 6 años (13). Dicha descripción coincide con un trabajo de España (8) en el que se observó que el déficit visual ocurrió después de la epilepsia y la ataxia fue el tercer síntoma de la enfermedad apareciendo a los 4 años junto con el inicio del deterioro cognitivo. Existe una variante finlandesa de la LCN infantil tardía (LCN 5) en la que las manifestaciones oculares son las más importantes, sin embargo, tienen un inicio de los síntomas más tardío entre los 4 y 7 años de edad, por lo que se tendría que acudir a técnicas moleculares de estudio genético para el diagnóstico definitivo (1). En 2 pacientes, casos 3 y 6 hubo confirmación por microscopia electrónica y análisis mutacional respectivamente. La edad de inicio fue tardío, a la edad de 4 años, en la niña que posteriormente se concluyó presentaba la forma Juvenil, o Enfermedad de Batten, correspondiente al caso 6, donde además de la edad de inicio el desarrollo de las crisis convulsivas fueron distintas, predominio de crisis parcial motora por más de 1 año, tiempo en el cual siguió acudiendo a clases, es decir el deterioro fue más lento, con el incremento de crisis y la presencia de otros tipos de convulsiones aparecen los demás componentes de regresión.
Las crisis epilépticas tónico clónicas generalizadas han sido descritas como el tipo de convulsión más frecuente (15). La presencia de múltiples tipos de crisis es un hallazgo frecuente en la LCN (16) lo que se observó en todos los casos analizados.
El diagnóstico diferencial en pacientes pediátricos con convulsiones mioclónicas en los que se han descartado malformaciones, infecciones y neoplasias conduce a la investigación de los diferentes tipos de Epilepsias Mioclónicas Progresivas (EMP) dentro de las cuales se encuentran las LCN que son el grupo más común de padecimiento neurodegenerativo de depósito lisosomal en la infancia (16).
Las crisis mioclónicas pueden ser de difícil reconocimiento, se manifiestan como sobresaltos breves generalizados o fragmentarios, varias veces en el día, se exacerban con estímulos auditivos y pueden ser fotosensibles (17,18). Debemos pues estar atentos ante la descripción que surja en el interrogatorio por parte de familiares o profesionales de la salud respecto a la semiología de crisis convulsivas menos frecuentes en la práctica diaria, como ausencias atípicas, mioclonías, astático-mioclónicas, espasmos infantiles y atónicas que se asocian a regresión del desarrollo (18).
Dentro de las Epilepsias Mioclónicas Progresivas y gracias a los estudios de genética molecular, han sido identificadas otras entidades además de la Lipofuscinosis como por ejemplo la Enfermedad de Lafora, Epilepsia Mioclónica de fibras rojas rasgadas, Enfermedad de Unverricht–Lundborg entre otras (16).
Los fármacos anticonvulsivantes deben seleccionarse teniendo en cuenta el tipo de crisis, el estado clínico del paciente y los efectos secundarios. Con frecuencia son necesarios varios fármacos para conseguir un adecuado control clínico, difícil de alcanzar en la mayoría de los pacientes formando éstas parte de las Epilepsias Refractarias. El valproato es uno de los medicamentos básicos en el tratamiento de la epilepsia de los pacientes con LCN. Es eficaz en las convulsiones generalizadas primarias y parciales secundariamente generalizadas (8). La lamotrigina puede ser preferida en mujeres debido a la teratogenicidad y a los efectos colaterales del valproato (19). Sin embargo se debe tener en cuenta que la lamotrigina puede empeorar las mioclonías (20). Levetiracetam y topiramato son altamente efectivos y son frecuentemente usados en combinación o como un tratamiento de segunda línea.
Algunos antiepilépticos pueden agravar las crisis mioclónicas (19). La carbamazepina, la fenitoina, gabapentina y la vigabatrina pueden empeorar las crisis mioclónicas por lo que están contraindicados (19,20,21).
Así pues la presencia de Epilepsia Mioclónica modifica la conducta médica: orienta hacia patologías específicas y determina la conducta farmacológica.
En todas las Resonancias Magnéticas de cráneo de los pacientes de nuestra serie se reprodujo el hallazgo esperado en esta entidad consistente en la presencia de atrofia cerebelosa y adelgazamiento de la corteza cerebral, se describe además en la literatura leve hiperintensidad de la sustancia blanca en T2. Estas alteraciones no son específicas y generalmente se correlacionan con la duración de la enfermedad (8). No presenta afectación de Ganglios Basales, característico de la forma Infantil tardía.
El hallazgo electroencefalográfico es precoz y característico en la LCN. Enlentecimiento progresivo de la actividad de base junto con la presencia de actividad epiléptica ictal e interictal, multifocal y generalizada, espigas, polipuntas, complejos punta onda lenta. La estimulación fótica a baja frecuencia 1-2 Hz manifiesta espigas de alto voltaje más de 200mV en regiones posteriores seguido de una onda lenta que corresponde a un potencial evocado gigante que es prácticamente una característica de esta patología (8).
Los Potenciales Evocados Visuales en fases precoces de la enfermedad, presenta respuestas gigantes a la estimulación luminosa intermitente. En el Electrorretinograma se extingue precozmente la respuesta por alteración de conos y bastones (2,8).
Hemos llegado a la sospecha diagnóstica uniendo los síntomas presentes (amaurosis adquirida con atrofia del Nervio Óptico, epilepsia con diferentes tipos de crisis, entre ellas resaltamos las mioclonías, pérdida de habilidades tanto motoras como cognitivas, atrofia supra e infratentorial, particularmente la atrofia de cerebelo es un dato a destacar para el diagnostico). El objetivo es certificar esta sospecha.
Los estudios de actividad enzimática (TPP1) preceden al estudio de microscopia electrónica cuando existe alta sospecha de enfermedad producida por mutación del gen CLN1, CLN2 (8). El poder medir la actividad enzimática de TTP1 y PPT1 respectivamente en muestras de sangre seca en papel calibrado permite el traslado de muestras y conservación a temperatura de ambiente de sitios lejanos a laboratorios especiales, pudiendo mantener su actividad enzimática hasta por 2 semanas (8,22).
Teniendo en cuentas los diferentes tipos de LCN, el screening incluye la identificación de inclusiones mediante la visualización al microscopio óptico de leucocitos o al microscopio electrónico de los tejidos biopsiados, la biopsia puede obtenerse de piel, conjuntiva, músculo esquelético. Los hallazgos más comunes son acumulación de lipopigmento autofluorescente en los lisosomas con variada ultraestructura (23,24). La búsqueda de linfocitos vacuolados y el análisis genético-molecular son métodos que se encuentran estandarizados en algoritmos diagnósticos (4).
Cabe destacar que en nuestro país no se cuentan con los métodos diagnósticos especializados que orienten hacia la mutación presente en las células o al déficit enzimático correspondiente por lo que la clínica se consideró, como en la mayoría de los casos, soberana y determinante.
El caso 7 se destaca por la manifestación conductual referida por el centro educativo al cual acudía, se trataba de una niña que presentaba convulsiones desde los 3 años, a los 5 años es evaluada en un centro de referencia de nuestro país indicando que desde los 4 años presentaba en la escuela dificultad en el relacionamiento, atención dispersa y cambios en el comportamiento. El cuadro clínico final de todos los pacientes consistió en epilepsia, deterioro motor, afectación visual y cognitiva con estado de dependencia total, sin embargo queremos hacer énfasis en el seguimiento del paciente desde la perspectiva del médico que ve a esta niña o niño en la fase inicial de la enfermedad, donde la convulsión está presente pero es el dato cognitivo-conductual el que inició toda la búsqueda etiológica. En otros como el caso índice el compromiso visual, sin embargo es la búsqueda de la unidad diagnóstica la que nos acerca al diagnóstico.
La evolución en el tiempo puede ser rápida hacia la regresión, como en el caso 8. El paciente puede fallecer en el primer episodio convulsivo o derivar a estado vegetativo permanente por años (1).
En todos los casos se constató presencia de síndrome pseudobulbar con indicación de gastrostomía. Entre ellos son 3 los fallecidos a la fecha del presente trabajo con edad promedio de defunción a los 12 años, y 3 sin seguimiento.
La presencia de insomnio y dolor no especificado en 3 pacientes en etapas avanzadas de la enfermedad y priapismo en un paciente nos hacen replantear respecto a la mirada que debemos tener hacia estos pacientes en estado de dependencia desde temprana edad, pero que sin embargo no están ajenos al dolor.
CONCLUSIONES
Frente a cada paciente el juicio clínico es la primera y fundamental herramienta. Definir el síntoma predominante y unir las diferentes áreas afectas de modo a establecer un hilo conductor en el proceso diagnóstico es el trabajo al cual estamos llamados. La LCN se debe plantear ante un paciente en la franja etaria promedio de 2 a 4 años que curse con regresión del desarrollo neurológico, epilepsia mioclónica y compromiso visual.
Consideramos que la certeza diagnóstica es siempre el objetivo ante todo paciente pero más aún en patologías hereditarias y progresivas con una evolución cierta a una muerte prematura.
La Lipofuscinosis ceroidea neuronal es una entidad aparentemente poco frecuente en nuestro medio, aunque consideramos que existe un subregistro en relación a las dificultades en el seguimiento, en establecer diagnóstico definitivo y a la ausencia de tratamiento que desalienta a las familias.
Inician con crisis convulsivas que no son diferentes por sí mismas al inicio del cuadro de tantas otras convulsiones que vemos en la infancia, es en el tiempo, siguiendo la evolución de estos pacientes que aparecen los elementos que conducen primero a plantear una epilepsia refractaria, posteriormente una Encefalopatia degenerativa ya con los componentes cognitivos, visuales, motores. El interrogatorio sobre el tipo de crisis cobra gran importancia, la Epilepsia Mioclónica Progresiva constituye un capítulo muy especial en la neuropediatría, la semiología de las convulsiones, constatar la presencia de diferentes tipos de crisis es un hallazgo que dirige la mirada hacia posibles etiologías.
La recurrencia en una misma familia se debe tener en cuenta, como hemos visto en la casuística presentada por lo que un adecuado consejo genético y contención por parte de los profesionales tratantes forma parte del cuidado de estos pacientes.
No existe aún un tratamiento definitivo para esta enfermedad. El manejo se orienta desde las fases iniciales a mejorar la calidad de vida del paciente y de su familia con medidas generales y de sostén. Los pacientes presentan complicaciones respiratorias, nutricionales e infecciosas que conducen a una situación aún más difícil de sobrellevar sin un apoyo social. Se están estudiando alternativas terapéuticas como la terapia génica consistente en introducir copias de genes mutados a través de virus que los insertarían en las células del cuerpo, esto sin embargo dista de tener resultados en un tiempo cercano, otra de las alternativas que generan mayores expectativas son las terapias de reemplazo enzimático, el problema se presenta por el escaso paso de niveles adecuados de enzimas a nivel cerebral, sin embargo, se encuentran en estudio diversas alternativas tales como modificaciones de ciertos carbohidratos presentes en las enzimas que podrían generar mayor disponibilidad y mejoría en los valores farmacocinéticos de difusión de dichas medicaciones (25).
Se deben generar políticas de salud que propicien la provisión de tecnologías adecuadas para el diagnóstico precoz de enfermedades genéticas como la descripta en este trabajo, además de impulsar la realización de estudios enzimáticos en Gota Seca, Biopsia de tejidos, estudios moleculares, neuroimágenes y electroencefalograma. El objetivo primario de todo esto es poder concluir un diagnóstico basado en una presunción clínica inicial.
El manejo interdisciplinario es una necesidad, lo cual exige la comunicación fluida entre los profesionales de modo a no generar más angustia en la familia. Es necesario enfrentar el desafío de dar la información de tal forma que dentro de lo humanamente posible la familia reciba el contenido pero también la contención.
REFERENCIAS
1. Méndez F, Mabe P. Lipofuscinosis ceroidea neuronal. Revista Pediátrica Electrónica [Internet]. 2005 Abril [citado 18 Nov 2013];2(1). [ Links ] Disponible en: http://www.revistapediatria.cl/vol2num1/12.htm
2. Goebel HH, Wisniewski KE. Current state of clinical and morphological features in human NCL. Brain Pathol. 2004;14:61-69. [ Links ]
3. Hobert JA, Dawson G. Neuronal ceroid lipofuscinoses therapeutic strategies: past, present and future. Biochimica et Biophysica Acta (BBA). Molecular Basis of Disease. 2006;1762(10):945-53. [ Links ] Available from: http://dx.doi.org/10.1016/j.bbadis.2006.08.004
4. Mole SE, Williams RE. Neuronal Ceroid-Lipofuscinoses. 2001 Oct 10 [Updated 2013 Aug 1]. In: Pagon RA, Adam MP, Bird TD, editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1428/ [ Links ]
5. Verma R, Raut TP, Tiwari N, Malhotra KP, Hussain N, Malhotra HS. Late infantile neuronal ceroid lipofuscinosis: a case report with review of literature. Ann Indian Acad Neurol [Internet]. 2013 [citado 2 Dic 2013];16:282-5. [ Links ] Disponible en: http://www.annalsofian.org/text.asp?2013/16/2/282/112500
6. Chang CH. Neuronal Ceroid Lipofuscinosis. eMedicine Website. [Last updated on 2013 Sep, Last accessed on 2013 Dec]. Available from: http://www.emedicine.com/neuro/topic498.htm [ Links ]
7. Orlin A, Sondhi D, Witmer M, Wessel M, Mezey J, Kaminsky S, et-al. Spectrum of Ocular Manifestations in CLN2-Associated Batten (Jansky-Bielschowsky) Disease Correlate with Advancing Age and Deteriorating Neurological Function. PLoS One [Internet]. 2013;8(8):e73128. Published online 2013 August 28. Available from: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0073128 [ Links ]
8. Pérez Poyato M. Espectro clínico-mutacional y estudios de correlación genotipo-fenotipo en la población española afectada de lipofuscinosis neuronal ceroidea [Tesis Doctoral]. Barcelona: Universitat de Barcelona; 2012. [ Links ] Disponible en: http://hdl.handle.net/2445/34740
9. Golabek AA, Kida E, Walus M, Wujek P, Mehta P, Wisniewski KE. Biosynthesis, glycosylation, and enzymatic processing in vivo of human tripeptidyl-peptidase I. J Biol Chem [Internet]. 2003 Feb 28;278(9):7135-45. [ Links ] (Cited 2013 Dec). Available from: http://hinari-gw.who.int/whalecomwww.ncbi.nlm.nih.gov/whalecom0/pubmed/12488460
10. Van Beersel G, Tihon E, Demine S, Hamer I, Jadot M, Arnould T. Different molecular mechanisms involved in spontaneous and oxidative stress-induced mitochondrial fragmentation in tripeptidyl peptidase-1 (TPP-1)-deficient fibroblasts. Biosci Rep [Internet]. 2013;33(2):e00023. [ Links ] Published online 2013 February 7. Prepublished online 2012 December 18. Available from:http://hinari-gw.who.int/whalecomwww.ncbi.nlm.nih.gov/whalecom0/pmc/articles/ PMC3566540/#__ffn_sectitle
11. Carvajal Cuenca A, Fernández Morales H, Cordero MH. Reporte de la primera familia costarricense con Neuropatía Óptica Hereditaria de Leber y una revisión del tema. Neuroeje [Internet]. 2006;20(1). [ Links ] (Citado en Dic 2013). Disponible en: http://www.binasss.sa.cr/revistas/neuroeje/20n1/art4.pdf
12. Pérez-Poyato MS, Pineda M, Ferrer I, Rodriguez-Revenga L, Cusi V, Martínez M, et-al. Late Infantile Neuronal Ceroid Lipofuscinosis: mutations in the CLN2 Gene and Clinical Course in Spanish Patient. J Child Neurol [Internet]. 2012. [ Links ] doi: 10.1177/0883073812448459. Available from: http://jcn.sagepub.com
13. Williams R, Vesa J, Jarvela I, McKay T, Mitchison H, Hellsten E, et-al. Genetic Heterogeneity in Neuronal Ceroid Lipofuscinosis: evidence That the Late-Infantile Subtype (Jansky-Bielschowsky Disease; CLN2) is not an allelic form of the Juvenile or Infantile Subtypes. Am J Hum Genet [Internet]. 1993;52:931-935. [ Links ] Available from: http://hinari-gw.who.int/whalecomwww.ncbi.nlm.nih.gov/whalecom0/pmc/articles/PMC1682401
14. La Morgia C. Rare mtDNA variants in Leber hereditary optic neuropathy families with recurrence of myoclonus. Neurology [Internet]. 2008 Mar 4 [citado dic 2010];70(10):762-70. [ Links ] Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18216301
15. Topeu M, Tan H, Yalnizoglu D, Usubutun A, Saatci I, Aynaci M, et-al. Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: clinical, neurophysiological, neuroradiological and histopathologic studies. Turk J Pediatr. 2004;46:1-10. [ Links ]
16. Andrade-Bañuelos A, Jean-Tron G, Ortega-Ponce F, Arnold S, Rana S, Islas-García D. Lipofuscinosis neuronal ceroide infantil tardia: reporte de un caso. Rev Mex Neuroci [Internet]. 2013;14(1):44-49. [ Links ] Disponible en: http://revmexneuroci.com/wp-content/uploads/2013/10/Nm131-09.pdf
17. Santavuori P, Haltia M. Infantile type of so-called neural ceroid-lipofuscinosis 1: a clinical study of 15 patients. J Neurol Sci. 1973;18:257-267. [ Links ]
18. Aicardi J, Plouin P, Goutiéres F. Ceroid-lipofuscinosis. Electroencephalogr Clin Neurophysiol. 1978;8:149-59. [ Links ]
19. Striano P, Belcastro V. Treatment of myoclonic seizures. Expert Rev Neurother [Internet]. 2012;12(12):1411-18. [ Links ] Available from: http://www.expert-reviews.com/doi/pdf/10.1586/ern.12.90
20. Syvertsen MR, Markhus R, Selmer KK, Nakken KO. Juvenile myoclonic epilepsy. Tidsskr Nor Laegeforen [Internet]. 2012 Aug 7;132(14):1610-3. [ Links ] Available from: http://tidsskriftet.no/article/2510744
21. Aberg L, Kirveskari E, Santavuori P. Lamotrigine therapy in juvenile neuronal ceroid lipofuscinosis. Epilepsia [Internet]. 1999 Jun;40(6):796-9. [ Links ] Available from: http://hinari-gw.who.int/whalecomwww.ncbi.nlm.nih.gov/whalecom0/pubmed/10368082
22. Miranda Contrerasa L, Delgado Luengoa W, Zerpab N, Chacín Hernándeza J, Chávezc CJ, González Ferrera S. Tripeptidil peptidasa 1 en pacientes con ceroidolipofuscinosis neuronal infantil tardía. An Pediatr (Barc). 2012;76(3):148-152. [ Links ]
23. Goebel HH. Morphologic diagnosis in neuronal ceroid lipofuscinosis. Neuropediatrics [Internet]. 1997;28:67-9. [ Links ] (Cited Dec 2013). Available from: http://hinari-gw.who.int/whalecomwww.ncbi.nlm.nih.gov/whalecom0/pubmed/915132
24. Arsenio-Nunes ML. Goutieres F. An ultramicroscopic study of the skin in the diagnosis of the infantile and late infantile types of ceroid-lipofuscinosis. J Neurol Neurosurg Psychiatry. 1975;38(10): 994-99. [ Links ]
25. Meng Y, Sohar I, Wang L, Sleat DE, Lobel P. Systemic Administration of Tripeptidyl Peptidase I in a Mouse Model of Late Infantile Neuronal Ceroid Lipofuscinosis: effect of Glycan Modification. PLoS One [Internet]. 2012;7(7):e40509. [ Links ] Published online 2012 July 6. Available from: http://hinari-gw.who.int/whalecomwww.ncbi.nlm.nih.gov/whalecom0/pmc/articles/PMC3391252/#__ffn_sectitle

