










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPediatría (Asunción)
On-line version ISSN 1683-9803
Pediatr. (Asunción) vol.40 no.2 Asunción Aug. 2013
CASO CLÍNICO
Síndrome de Jarcho- Levin. Disostosis espondilocostal. Reporte de dos Casos Clínicos
Jarcho-Levin Syndrome (Spondylocostal Dysostosis): A Report of Two Clinical Cases
María Beatriz NP de Herreros, Rubén Franco, Oscar Atobe(1).
1. Secretaria Nacional de Discapacidad- SENADIS. Asunción. Paraguay.
Correspondencia: Dra. María Beatriz NP de Herreros. E-mail: maranp-4@hotmail.com
Recibido: 30/05/2013; Aceptado: 26/06/2013.
RESUMEN
Introducción: El síndrome de Jarcho-Levin incluye un conjunto de fenotipos clínicos caracterizados por enanismo y tronco corto con múltiples anomalías de segmentación de las vertebras y anomalías costales. Existen dos fenotipos que se distinguen por sus características clínicas, radiológicas, su tipo de herencia, y su pronóstico: la disostosis espondilotorácica y la disostosis espondilocostal. En la primera se observa anomalía de los cuerpos vertebrales y de costillas, en un patrón de aspas de ventilador. En la disostosis espondilocostal se ven malformaciones de vértebras y costillas, pero las costillas no se disponen en ventilador y suele ser más leve. Casos Clínicos: El caso clínico 1 corresponde a una niña de 8 días de vida con múltiples malformaciones de vértebras y costillas, sin otros defectos, y el segundo, es una niña de 33 días con múltiples defectos de vértebras y costillas, sin otras malformaciones. En este caso se realizó el diagnóstico prenatal por ecografía. No se han encontrado antecedentes familiares ni se ha observado dificultad respiratoria, y ambas niñas están evolucionando muy bien. Discusión: La disostosis espondilocostal es una patología genética rara de origen autosómico recesivo y el diagnóstico temprano y certero favorece la buena evolución del paciente y permite realizar el asesoramiento genético.
Palabras clave: Disostosis espondilocostal, disostosis espondilotorácia, Jarcho Levin, anomalías vertebrales, anomalías costales.
ABSTRACT
Introduction: Jarcho- Levin syndrome includes a series of clinical phenotypes characterized by dwarfism, short trunk and multiple vertebral segmentation defects and anomalies of the ribs. There are two phenotypes with different clinical and radiological characteristics, different mode of heredity and different prognosis; the spondylothoracic dysostosis and the spondylocostal dysostosis. The first one shows multiple anomalies of vertebral bodies and ribs and a short thorax in a“fan like” pattern, the second one shows the same anomalies, but not in a fan pattern and it is usually milder. Cases report: We report two cases of spondylocostal dysostosis. The first case is a girl of 8 days of age with multiple anomalies of vertebral bodies and ribs and no other defects. The second case a girl of 33 days of age with multiple anomalies of vertebral bodies and ribs without any other malformations. The latter case had prenatal diagnosis in the second trimester of the pregnancy. There was no family history in any of the two cases, and they don´t have respiratory distress. Both girls are evolving very well. Conclusion: The spondylocostal dysostosis is a rare genetic autosomal recessive syndrome. Early and accurate diagnosis is very important for the good evolution of the patient and allows good and precise genetic counseling.
Key words: Spondylocostal dysostosis, spondylothoracic dysostosis, Jarcho Levin, vertebral anomalies, rib anomalies.
INTRODUCCIÓN
En 1938 Saul Jarcho y Paul Levin del Hospital John Hopkins, reportaron varios casos de insuficiencia respiratoria debida a anomalías vertebrales y costales. Casi 30 años después en 1966 Norman Lavy describió un síndrome similar en una familia de Puerto Rico y luego John Mosely también reportó el síndrome y utilizó por primera vez el nombre de Displasia Espondilotorácia(1).
El síndrome de Jarcho-Levin es un epónimo que incluye un conjunto de fenotipos clínicos caracterizados por enanismo y tronco corto con múltiples anomalías vertebrales y costales. Existen dos fenotipos incluidos bajo este epónimo que se distinguen por sus características clínicas y radiológicas, su tipo de herencia, y su pronóstico: la Disostosis espondilotorácica y la Disostosis espondilocostal(1-5).
La prevalencia de este síndrome ha sido estimada en 1 en 12.000 nacidos vivos, en la población de Puerto Rico. No hay datos de prevalencia para el resto del mundo, pero se calcula que es de 1 en 200 000 recién nacidos, o sea, muy raro(3). Este síndrome es de origen genético y han sido identificadas varias mutaciones en relación al mismo. Las diferentes mutaciones genéticas encontradas son: SCDO1, que es una forma de disostosis espondilocostal que está causada por una mutación en el gen DLL3, localizado en el cromosoma 19q13. Otras formas de SCDO incluyen a la SCDO2, causada por una mutación en el gen MESP2 en el cromosoma 15q26.1, la SCDO3, causada por la mutación en el gen LFNG en el cromosoma 7p22 y la SCDO4, causada por la mutación en el gen HES7 en el cromosoma 17p13.2. También ha sido descrita una forma dominante de SCDO y esta es llamada la SCDO5(1).
Por más de 50 años ha habido confusión con respecto a la diferenciación de los dos síndromes que causan insuficiencia respiratoria. La Disostosis Espondilocostal (SCDO) o Síndrome de Jarcho Levin causa insuficiencia respiratoria leve a moderada y es pan-étnica y la Disostosis Espondilotorácica (DET) o síndrome de Lavy-Mosely produce mayor dificultad respiratoria, y se ve en los portorriqueños(3). Se ha tratado de establecer varias clasificaciones del síndrome de Jarcho-Levin según los diferentes grados de severidad, de los defectos costovertebrales, y la presencia de otros defectos. De hecho, y a pesar de la gran heterogeneidad clínica y etiológica, se siguen incluyendo todos los tipos juntos(6). Las características clínicas consisten en talla corta de presentación prenatal, tórax y cuello cortos, abdomen protuberante, occipucio prominente, puente nasal ancho, narinas antevertidas, fisuras palpebrales de inclinación mongoloide. También se pueden ver pectus carinatum, aumento del diámetro antero-posterior del tórax, cifo-escoliosis y lordosis. Los miembros son normales, aunque impresionan más largos. A la exploración radiográfica se observan múltiples defectos de segmentación de vertebras y costillas(1-7). Se pueden ver anomalías asociadas como anomalías del tubo neural, hernias inguinales y/o umbilicales, anomalías del tracto urinario y anomalías cardiacas. También se pueden ver otras malformaciones como; hernia diafragmática, paladar hendido, criptorquidia, y arteria umbilical única(2-16).
CASOS CLÍNICOS
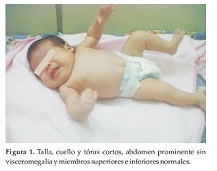
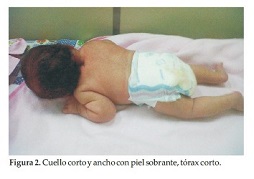
Caso Clínico 1. Niña de 33 días que consulta por talla y cuello cortos. La niña es el producto del primer embarazo de una pareja joven, aparentemente sana y no consanguínea. No hay antecedentes familiares, ni de patologías o ingestión de medicamentos durante el embarazo. El diagnóstico prenatal se realizó por ecografía en el segundo trimestre del embarazo. La niña nace en un parto por cesárea, a las 39 semanas de gestación, con peso de 3170 grs. (P 25), talla de 43 cm (perc < 3) y CC: 34, 5 cm (P 50). En el momento de la consulta tiene 33 días de vida. Examen Físico: Talla: 48, 5 cm (P 10), CC: 34, 5 cm (P 10) y Peso: 4200 grs. (P 50). Al examen físico la niña presenta (Figura 1). Talla, cuello y tórax cortos; abdomen prominente sin visceromegalia y miembros superiores e inferiores normales (Figura 2). Cuello corto y ancho con piel sobrante. Los genitales y los ruidos cardiacos son normales.
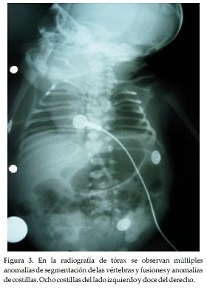
Los estudios realizados fueron: Test del piecito: normal. Emisiones otoacústicas: células ciliadas que no han completado su desarrollo. Ecografía renal: normal. Ecoencefalografía: Normal. En los controles posteriores, la niña a los 6 meses sigue en buen estado de salud sin presentar dificultad respiratoria. En la radiografía de tórax se observan múltiples anomalías de segmentación de las vértebras, fusiones y anomalías de costillas (Figura 3).
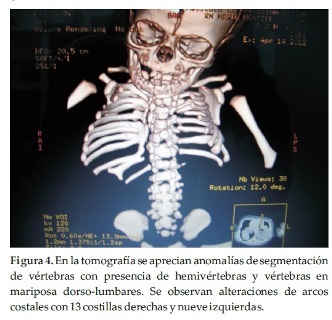
En la tomografía se aprecian anomalías de segmentación de vértebras con presencia de hemivértebras y vértebras en mariposa dorso-lumbares. Se observan además alteraciones de los arcos costales con trece costillas derechas y nueve izquierdas, con presencia de fusión proximal del noveno y décimo arcos costales posteriores y ensanchamiento de aspecto bífido de segmento distal de la duodécima costilla derecha (Figura 4).
Se realiza el diagnóstico de Disostosis Espondilocostal o Síndrome de Jarcho Levin, de gravedad moderada y se procede al asesoramiento genético familiar dando un riesgo de recurrencia del 25%, por ser considerada una patología genética de origen génico autosómico recesivo.
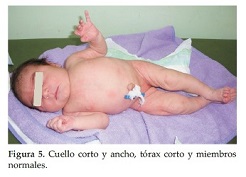
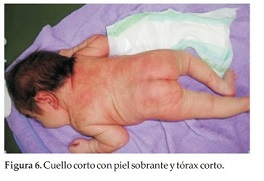
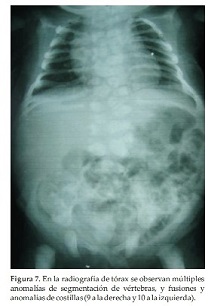
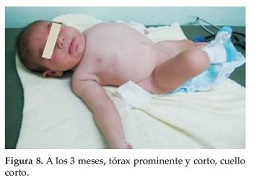
Caso Clínico 2. Niña de 8 días de vida que consulta por talla, tórax y cuello cortos. La niña es el producto del primer embarazo de una pareja joven, aparentemente sana y no consanguínea, sin antecedentes familiares, antecedentes patológicos del embarazo actual, ni de ingestión de medicamentos. No se realizó el diagnóstico prenatal, la madre no se hizo ecografías durante el embarazo. La niña nace a las 38 semanas en un parto normal, con peso de 3450 grs. (P 50), talla: 45 cm. (perc:< 3) y CC: 32, 5 cm (P 10). En la primera consulta tiene 8 días de vida. Examen Físico: Talla: 45 cm (perc < 3), peso: 3250 grs. (P 25) y CC: 33 cm. (P 25). Talla, tórax y cuello cortos. Abdomen prominente sin visceromegalia. Genitales y miembros superiores e inferiores normales. Ruidos cardiacos normales. Cuello corto y ancho, tórax corto y miembros normales(Figura 5). Cuello corto con piel sobrante y tórax corto (Figura 6). En la radiografía de tórax se observan múltiples anomalías de segmentación de vértebras, y fusiones y anomalías de costillas (9 a la derecha y 10 a la izquierda) (Figura 7). A los 3 meses, tórax prominente y corto, cuello corto (Figura 8).
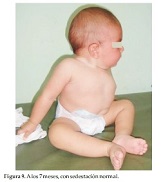
A los 7 meses con sedestación normal (Figura 9). La niña sigue en buen estado de salud, con desarrollo sicomotor normal y no presenta dificultad respiratoria. En este caso también se realiza el diagnóstico de Disostosis Espondilocostal o Síndrome de Jarcho- Levin y se procede al asesoramiento genético, dando un riesgo de repetición del 25%. La comparación de los dos casos se puede observar en la tabla 1.

DISCUSIÓN
El Síndrome de Jarcho- Levin o Disostosis espondilocostal es un trastorno de etiología genética que se caracteriza por la presencia de múltiples anomalías vertebrales y costales. Estas anomalías pueden estar asociadas o no, a otros defectos. La patogénesis de estas malformaciones ocurre en las seis primeras semanas de vida intrauterina durante el proceso de diferenciación mesenquimal. Las anomalías costales, aparentemente, son consecuencia de las anomalías vertebrales y suelen localizarse con mayor frecuencia en la concavidad de la curva escoliótica. Ya que la patología ocurre intraútero, sería importante llegar al diagnóstico en la etapa prenatal, en el segundo o tercer trimestre del embarazo. A pesar de la característica de trasmisión hereditaria de este síndrome, en ninguna de nuestras pacientes hemos encontrado antecedentes familiares. El principal problema médico de estos pacientes se presenta en el aparato respiratorio, ya que presentan insuficiencia respiratoria de tipo restrictivo, con infecciones a repetición. Es de gran importancia el tratamiento agresivo de estas infecciones, la consulta rápida y la internación preventiva. En la mayoría de los casos, el tratamiento de las malformaciones es conservador, con controles radiográficos periódicos, fisioterapia y control de las infecciones.
En las escoliosis producidas por esta patología, está demostrada la inefectividad de los corsés ortopédicos y en los casos con grandes deformidades progresivas se requiere el tratamiento quirúrgico para la corrección de la escoliosis y/o de las deformidades costales. El diagnóstico de certeza y la clasificación apropiada de los diferentes fenotipos ayudará a mejorar el asesoramiento genético con respecto al manejo, pronóstico y riesgo de recurrencia en la familia. Nuestras dos pacientes están presentando una evolución muy favorable, sin complicaciones respiratorias hasta el momento. En ambos casos se realizó el asesoramiento genético familiar dando un riesgo de repetición del 25%, por ser una patología génica de origen autosómico recesivo(1,3,5,16-18).
REFERENCIAS
1. Mc Kusick. On Line Mendelian Inheritance in man. OMIM. 277300 [ Links ]
2. Cornier AS, Ramirez N, Carlo S, Reiss A. Controversies surrounding Jarcho-Levin syndrome. Curr Opin Pediatr. 2003;15(6):614-20. [ Links ]
3. Cornier AS, Staehling-Hampton K, Delventhal KM, Saga Y, Caubet JF. Mutations in the MESP2 gene cause spondylothoracic dysostosis/Jarcho-Levin syndrome. Am J Hum Genet. 2008;82(6):1334-41. [ Links ] Epub 2008 May 15.
4. Cornier AS. Spondylothoracic Dysostosis. 2010 Aug 5. In: Pagon RA, Adam MP, Bird TD, editors. GeneReviews (Internet). Seattle (WA): University of Washington, Seattle; 1993-2013. [ Links ] Available from: http://www.ncbi.nlm.nih.gov/books/NBK45316
5. Freire-Abelleira C, González-Herranz P, De la Fuente-González C, Castro-Torre M. Anomalías congénitas de vértebras y costillas: síndrome de Jarcho-Levin: revisión clínica.Acta Ortop Gallega. 2006;2(1):7-10. [ Links ]
6. Martínez-Frías ML, Bermejo Sánchez E, Martínez Santana S, Nieto Conde C, Egüés Jimeno J, Pérez Fernández JL, et al. Síndromes de Jarcho-Levin y Casamassima: diagnóstico diferencial y frecuencia en España. Anales Españoles de Pediatría. 1998;48(5):512-14. [ Links ]
7. Jones KL. Recognizable Patterns of Human Malformation. 6th ed. USA: Elsevier Saunders; 2006. [ Links ]
8. Onay OS, Kinik ST, Otgun Y, Arda IS, Varan B. Jarcho-Levin syndrome presenting with diaphragmatic hernia. Eur J Pediatr Surg. 2008;18(4):272-74. [ Links ] Epub 2008 Jul 15.
9. Day R, Fryer A. Diaphragmatic hernia and preaxial polydactyly in spondylothoracic dysplasia. Clin Dysmorphol. 2003;12(4):277-78. [ Links ]
10. Swietlinski J, Swist-Szulik K, Maruniak-Chudek I, Pyrkosz A. Spondylothoracic dysostosis associated with diaphragmatic hernia and camptodactyly. Genet Couns. 2002;13(3):309-17. [ Links ]
11. Duru S, Ceylan S, Güvenç BH. Segmental costovertebral malformations: association with neural tube defects. Report of 3 cases and review of the literature. Pediatr Neurosurg. 1999;30(5):272-27. [ Links ]
12. Simpson JM, Cook A, Fagg NL, MacLachlan NA, Sharland GK. Congenital heart disease in spondylothoracic dysostosis: two familial cases. J Med Genet. 1995;32(8):633-35. [ Links ]
13. Hatakeyama K, Fuse S, Tomita H, Chiba S. Jarcho-levin syndrome associated with a complex congenital heart anomaly. Pediatr Cardiol. 2003;24(1):86-88. [ Links ]
14. Dane B, Dane C, Aksoy F, Cetin A, Yayla M. Jarcho-Levin syndrome presenting as neural tube defect: report of four cases and pitfalls of diagnosis. Fetal Diagn Ther. 2007;22(6):416-19. [ Links ] Epub 2007 Jul 24.
15. Etus V, Ceylan S, Ceylan S. Association of spondylocostal dysostosis and type I split cord malformation. Neurol Sci. 2003;24(3):134-37. [ Links ]
16. Adegboyega PA, Adesokan AA, Sample TG, Nichols MM. Pathological case of the month. Spondylothoracic dysplasia with multiple congenital cardiac anomalies. Arch Pediatr Adolesc Med. 1996;150(2):221-22. [ Links ]
17. Beine O, Bolland J, Verloes A, Lebrun FR, Khamis J, Muller Ch. Spondylocostal dysostosis: a rare genetic disease. Rev Med Liege. 2004; 59(9):513-16. [ Links ]
18. Karnes PS, Day D, Berry SA, Pierpont ME. Jarcho-Levin syndrome: four new cases and classification of subtypes. Am J Med Genet. 1991;40(3):264-70. [ Links ]

