










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPediatría (Asunción)
On-line version ISSN 1683-9803
Pediatr. (Asunción) vol.39 no.1 Asunción Apr. 2012
ARTÍCULO ORIGINAL
Detección de la deleción F508 del gen CFTR por la técnica de mutagénesis dirigida mediante PCR en pacientes con Enfermedad Fibroquística
Detection of F508 Deletion in the CFTR Gene by PCR Directed Mutagenesis in Patients with Fibrocystic Disease
Marta Ascurra(1), María Celeste Vega-Gómez(2), José L. San-Millán(3), Dolores Tellería(3), Andrés Mojoli Le Quesne(2), María José Fernández-Nestosa(4)
1. Programa de Prevención de la Fibrosis Quística y del Retardo Mental del Ministerio de Salud Pública y Bienestar Social. Asunción, Paraguay.
2. Centro para el Desarrollo de la Investigación Científica (CEDIC). Asunción, Paraguay.
3. Unidad de Genética Molecular, Hospital Universitario Ramón y Cajal, e Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Madrid, España.
4. Facultad Politécnica. Universidad Nacional de Asunción (FP-UNA). San Lorenzo, Paraguay.
Correspondencia: María José Fernández-Nestosa. Laboratorio de Computación Científica y Aplicada (LCCA). FP-UNA. Campus Universitario UNA, San Lorenzo, Paraguay. E-mail: mjfernandez@pol.una.py
Recibido: 24/02/2012, aceptado para publicación: 29/03/2012.
RESUMEN
La Fibrosis quística es la enfermedad autosómica recesiva más común en la población blanca y se caracteriza por la obstrucción de conductos, principalmente en pulmón, páncreas y tracto genital. Se presenta en uno de cada 2000 a 2500 nacidos vivos y tiene una frecuencia de portadores de uno cada 20 a 25 nacidos vivos. La enfermedad es causada por diferentes mutaciones en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR). La mutación más frecuente en el gen CFTR es la deleción de tres pares de bases (CTT) denominada F508. Este trabajo se realizó con el fin de estandarizar la técnica de mutagénesis dirigida mediante PCR (PSM) para la detección de.la mutación F508 en pacientes con fibrosis quística. El método utilizado fue validado mediante secuenciación del DNA del exón 10 en todos los individuos. Mediante este análisis genético se detectaron seis individuos con las mutaciones F508 e I507. El método implementado en nuestro laboratorio podría servir para realizar un sondeo poblacional de portadores de mutaciones para la FQ.
Palabras clave: Fibrosis quística, F508, PSM, sitio de restricción.
ABSTRACT
Cystic fibrosis is the most common autosomal recessive disease in the Caucasian population and is characterized by obstruction of passages, especially of the lungs, pancreas, and genital tract. It presents in 1 of every 2000 to 2500 live births, and 1 of every 20 to 25 newborns are carriers. The disease is caused by different mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The most common mutation in the CFTR gene is the deletion of three base pairs (CTT) known as F508.Our study was performed for the purpose of standardizing PCR-mediated site-directed mutagenesis (PSM) for detection of the F508 mutation in patients with cystic fibrosis. The method used was validated by DNA sequencing of exon 10 in all individuals. This genetic analysis detected 6 persons with F508 and I507 mutations. The method used in our laboratory could be useful for carrying out a population survey for carriers of the mutations for cystic fibrosis.
Keywords: Cystic fibrosis, F508, PSM, restriction site.
INTRODUCCIÓN
La fibrosis quística (FQ) es la más común de las enfermedades autosómicas recesivas en la población de raza blanca. Se presenta en uno de cada 2.000- 2.500 nacidos vivos y tiene una frecuencia de portadores de uno cada 20-25 nacidos vivos (1). Se caracteriza clínicamente por la obstrucción de los conductos en el pulmón, páncreas y tracto genital, principalmente debido a que el tejido epitelial de los órganos afectados se resiente por su incapacidad de transportar eficientemente el cloruro a través de la membrana celular (2-4).
El gen relacionado con la enfermedad reside en el cromosoma 7 q31-32 y codifica para una proteína llamada proteína reguladora de la conductancia transmembrana de la FQ (CFTR). Las mutaciones en el gen CFTR resultan en la falta de proteína funcionalmente activa. La más frecuente es la deleción F508, pero se han descrito más de 1500 mutaciones posibles que varían ampliamente según el origen étnico y la localización geográfica de cada población (5). La deleción F508 consiste en una deleción de 3 pares de bases (CTT) en el exón 10, que resulta en la pérdida de un aminoácido (fenilalanina) en la posición 508 de la proteína (6). Como consecuencia de esta deleción se genera una proteína CFTR que no se pliega de manera normal y acaba siendo degradada por la célula (7,8).
La frecuencia de la deleción F508 varía entre un 70% en países del norte de Europa y valores cercanos al 50% en España y países limítrofes como Argentina. Debido al alto porcentaje de mutaciones que cubre la deleción F508, más del 50% de los cromosomas fibroquísticos (5), y a la falta de diagnóstico molecular en los pacientes que padecen enfermedad fibroquística en el Paraguay, es de gran importancia detectar esta alteración como apoyo al diagnóstico clínico y también como única vía de detección de portadores. El conocimiento del genotipo es también de utilidad para la predicción de ciertas características fenotípicas, como la función pancreática (correlación genotipo-fenotipo) y la categorización de pacientes para el diseño e implementación de futuras estrategias terapéuticas (9).
Nuestro propósito fue implementar una técnica que permita la detección de la mutación F508, para luego aplicarla al diagnóstico de posibles afectados y sus familiares. La metodología empleada se denomina mutagénesis dirigida mediante PCR (PSM), fue desarrollada por Roqué y colaboradores (10) y se basa en la posibilidad de introducir mutaciones puntuales durante la reacción de PCR. Los cambios de secuencia se eligen de modo que generen sitios de reconocimiento para enzimas de restricción sólo en un alelo específico. La técnica fue introducida por Haliassos y colaboradores (11) en Francia para la rápida detección de mutaciones en el oncogén ras y fue aplicada a la FQ por Friedman y colaboradores (12,13).
La estrategia empleada consistió en utilizar un cebador 5’ con un cambio puntual en una base que linda con la mutación F508. Este cambio introduce un sitio de corte para la enzima Mbol (5’GATC3’) en el alelo sano, que no aparece en el alelo con la deleción F508 por no completarse la secuencia de corte. Para el extremo 3’ se utiliza un cebador que permite abarcar en el producto de amplificación un sitio de corte constitutivo que sirve como control interno de la actividad de la enzima de restricción. La implementación de esta metodología nos permitió detectar portadores para las mutaciones F508 y I507.
MATERIAL Y MÉTODOS
Población: Para la puesta a punto de la metodología se obtuvo el consentimiento de siete pacientes con fibrosis quística del Programa de Prevención de la Fibrosis Quística y del Retardo Mental, Ministerio de Salud Pública y Bienestar Social (MSPyBS). En los casos de menores de edad, el consentimiento fue dado por sus padres. Como criterio de inclusión se consideró a posibles portadores de la mutación F508 con un claro diagnóstico clínico de fibrosis quística.
Diagnóstico Genético: Se obtuvo ADN genómico a partir de sangre total extraída con EDTA como anticoagulante utilizando el kit Qiamp DNA Blood Mini (Qiagen). El ADN resuspendido en agua se amplificó con los cebadores CFTR Fw y CFTR Rv (Figura 1). El programa de amplificación se realizó en un termociclador con 25-40 ciclos (1 minuto: 94°C, 2 minutos: 50°C, 1 minuto, 72°C).

Digestión enzimática: Para la digestión enzimática, se incubó una alícuota del producto de amplificación con la enzima de restricción MboI (New England Biolabs). Las muestras se corrieron en un gel de agarosa NuSieve 3:1 (Lonza) al 4%. Los geles fueron visualizados con el reactivo EZ-Vision (Amresco) bajo luz ultravioleta.
Secuenciación de DNA: Con el fin de comprobar la validez del método utilizado, el DNA amplificado por PCR del exón 10 de todos los individuos fue secuenciado en un laboratorio homologado para el estudio molecular del gen CFTR (Hospital Ramón y Cajal, Madrid).
RESULTADOS
En una primera etapa, el objetivo fue estandarizar la técnica utilizando ADN de un individuo sano y un portador de la mutación F508. Se determinaron las condiciones óptimas para la amplificación del exón 10 del gen CFTR por PCR, los productos sin cortar se muestran en la Figura 2a. Se puede observar claramente una banda correspondiente al exón 10 del gen CFTR en ambos individuos (S y P1). El tamaño de la banda es de 262 pb (pares de bases) en el caso del alelo sano y de 259 pb en el alelo con la mutación F508. Sin embargo, el tamaño del gel y las condiciones de corrida no son suficientes para detectar esta pequeña diferencia. Luego del corte con la enzima de restricción, todas las bandas disminuyen de tamaño, indicando que la enzima ha cortado el 100% del producto de amplificación. En la Figura 2b se puede comprobar que las bandas correspondientes al alelo sano y al mutado se separan, dando dos bandas ahora diferenciables. Los alelos con la deleción F508 no presentan sitio de corte para Mbol y, por lo tanto, migran por encima del alelo sano. El tamaño de los fragmentos después del corte es de 201 pb en el caso del alelo normal y 215 pb en el del alelo mutado.
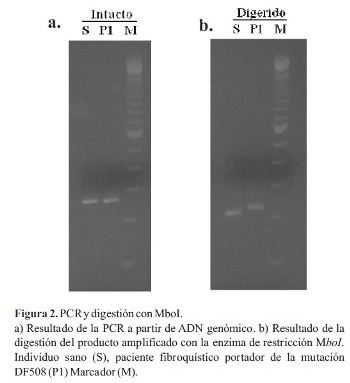
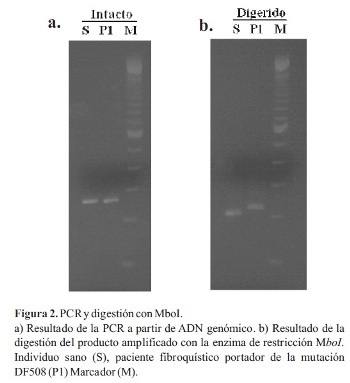
Una vez puesto a punto el método, en una segunda etapa, se analizó el ADN de otros seis pacientes fibroquísticos (P2-P7). Se realizó la extracción ADN de todos los individuos y se procedió a realizar la detección de la mutación mediante PSM. En el gel de diagnóstico (Figura 3) se puede observar que los pacientes P1, P3, P4 y P6 muestran un patrón característico de individuo homocigota para la mutación F508. El paciente P2 presentó un patrón idéntico al control sano (S), lo cual sugiere que no presenta la mutación buscada en ninguno de los dos alelos del gen CFTR. En el caso de los pacientes P5 y P7 se observó un patrón correspondiente a un heterocigota para la mutación F508, con un alelo mutado de mayor tamaño y un alelo sin la mutación que migra igual que el control sano.

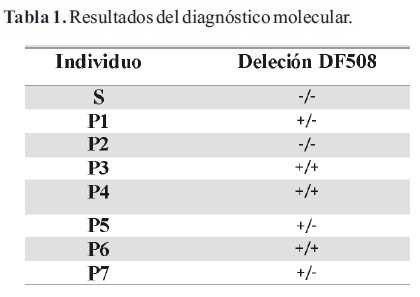
Los resultados de la secuenciación de exón 10 del gen CFTR confirmaron los obtenidos por PSM, a excepción del paciente P1 (Tabla 1). Dicho individuo presenta patrón correspondiente a un homocigota para mutación F508 según la técnica de PSM, mientras que los resultados de la secuenciación demostraron que se trata de un heterocigota F508/I507. Mediante el análisis de la secuencia de los mutantes F508 y I507 se comprobó que la estrategia utilizada no permite discriminar las mutaciones F508 y I507 (Figura 4).

DISCUSIÓN
Dado que hay tantas mutaciones distintas registradas en el gen CFTR, es de gran utilidad la existencia de una alta diversidad de técnicas para el análisis genético. El método empleado en el presente estudio es pues una alternativa a otros métodos costosos y complejos que requieren corridas en geles de alta resolución o hibridación con oligoalelos específicos para la detección de mutaciones. La técnica de PSM tiene la ventaja de poder ser aplicada no sólo para la detección de esta deleción sino también para cualquier otro tipo de mutación presente en este gen.
Sobre la base de los resultados obtenidos, se podría deducir que 6 de los 7 pacientes analizados son portadores de la deleción F508. De entre las 14 posibles mutaciones se detectaron 9 correspondientes a la F508, una cifra que se corresponde con lo que cabría esperar teniendo en cuenta que más del 50% de los cromosomas fibroquísticos tienen la mutación DF508. Es importante aclarar, sin embargo, que esto no pretende ser un estudio de frecuencia de la mutación F508 en la población paraguaya, ya que se han seleccionado fenotipos graves y es lógico que se detecten mutaciones de clara influencia en el fenotipo.
En los resultados obtenidos se comprobó que la técnica desarrollada por Roqué y colaboradores(10) permite discriminar el alelo sano del alelo con la mutación F508. Sin embargo, esta estrategia no es específica para la identificación de la mutación F508, tal como se comprobó en el caso de individuo heterocigota F508/I507. Por esta razón, se propone la incorporación de una segunda prueba, específica para la mutación I507, sólo para aquellos individuos positivos para mutación F508 (Figura 5). La estrategia consiste en utilizar un cebador 5’ con un cambio puntual en una base que linda con la mutación I507.

Este cambio introduce un sitio de corte para la enzima BglII (5’AGATCT3’) en el alelo con la deleción I507que no aparece en el alelo con la deleción F508 ni en el alelo sano. De esta forma, utilizando otra variante de la técnica de PSM, se podría discriminar entre las mutaciones F508 e I507. Es importante destacar que el problema de la falta de discriminación se vería minimizado por la gran diferencia, del orden de 50 veces, de frecuencia entre ambas mutaciones en otras poblaciones.
En aquellos individuos en que el método utilizado no detectó la presencia de dos mutaciones, se procedió a hacer un barrido en 17 de los 27 exones del gen CFTR, en los que se ha detectado más frecuentemente mutaciones FQ, mediante la técnica de DHPLC (Denaturing High Performance Liquid Chromatography)(14). En el individuo P7, heterocigoto para F508, se detectó la presencia de la mutación 4005+1G>A, situada en el primer nucleótido del exón 20. Esta mutación se ha detectado, con baja frecuencia, en diferentes poblaciones europeas, y cuando aparece en combinación con F508del, provoca un fenotipo grave.
A fin de definir el espectro mutacional de la FQ en nuestro medio, sería de mucha importancia realizar análisis poblacionales de tipos y frecuencias mutacionales presentes en los pacientes fibroquísticos de Paraguay. Además, sería necesario realizar un sondeo poblacional de portadores de las mutaciones más frecuentes que permitiría definir mejor el riesgo real de FQ en la población. Estos datos permitirían pronosticar con mayor exactitud los riesgos de herencia y desarrollo de la enfermedad en familias fibroquísticas. La técnica puesta a punto en nuestro laboratorio serviría para realizar un sondeo poblacional de portadores de mutaciones para la FQ, paso siguiente a encarar por nuestro equipo.
Sería muy útil poder continuar con los estudios de otras mutaciones del gen CFTR en estos mismos pacientes para poder correlacionar los hallazgos clínicos con las diferentes combinaciones asociadas de otras mutaciones frecuentes en la región como ser G542X, R553X, W1282X, N1303K, R117H, entre otras.
AGRADECIMIENTOS
Agradecemos a las Doctoras Antonieta Rojas de Arias y Leticia Olavarrieta Sccapini por sus sugerencias y por la revisión de este manuscrito, a los Licenciados Ana Gómez y a Jorge Alfonso por la asistencia técnica. Este trabajo fue financiado por la Dirección General de Investigación Científica y Tecnológica de la Universidad Nacional de Asunción (UNA). Las muestras de los pacientes con FQ fueron brindadas por la Dra. Marta Ascurra, Directora del Programa de Prevención de la Fibrosis Quística y del Retardo Mental del Ministerio de Salud Pública y Bienestar Social (MSPYBS).
REFERENCIAS
1. Chertkoff L. Anales de la Fundación Alberto Roemmers. Buenos Aires: Roemmers; 1998. [ Links ]
2. Gelehrter TD, Collins FS. Principles of the medical genetics. Baltimore: Williams & Wilkins; 1990. [ Links ]
3. Collins FS. Cystic fibrosis: molecular biology and therapeutic implications. Science. 1992; 256:774-79. [ Links ]
4. Welsh MJ, Smith AE. Fibrosis quística. Investigación y Ciencia. 1996;233:16-24. [ Links ]
5. Cystic Fibrosis Genetics Analysis Consortium. Population variation of common cystic fibrosis mutations. Hum Mutat. 1994;4:167-77. [ Links ]
6. Kerem B, Rommens JM, Buchanan JA. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073-1080. [ Links ]
7. Cheng SH, Gregory RJ, Marshall J. Defective intracellular traffic and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827-34. [ Links ]
8. Bear CE, Li CH, Kartner N. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell. 1992;68:809-18. [ Links ]
9. Paraguay. Ministerio de Salud Pública y Bienestar Social. Dirección General de Programas de Salud. Programa de Prevención de Fibrosis Quística y del Retardo Mental. Guía Clínica: Fibrosis Cística. Asunción:MSPBS; 2009. [ Links ]
10. Roque M, Castellanos M, Pott-Godoy C. Diagnóstico de la deleción DF508 en el gen CFTR a través de mutagénesis dirigida mediada por PCR. Arch Argent Pediatr. 2000;98(5): 304-309. [ Links ]
11. Haliassos A, Chomel JC, Tesson L. Modification of enzymatically amplified DNA for the detection of point mutations. Nucleic Acids Res. 1989;17:3606. [ Links ]
12. Friedman KJ, Highsmith EW, Silverman LM. Detecting multiple cystic fibrosis mutations by polymerase chain reaction-mediated site-directed mutagenesis. Clin Chem. 1991;37:753-55. [ Links ]
13. Friedman KJ, Highsmith EW.Jr. Molecular biology and pathology. London: Academic Press; 1993. [ Links ]
14. Le-Marechal C, Audrezet MP, Quere I, Raguenes O, Langonne S, Ferec C. Complete and rapid scanning of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by denaturing highperformance liquid chromatography (D-HPLC): major implications for genetic counselling. Hum Genet. 2001;108:290-98. [ Links ]

