










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPediatría (Asunción)
versão On-line ISSN 1683-9803
Pediatr. (Asunción) v.38 n.3 Asunción dez. 2011
CASO CLÍNICO
Síndrome de Adams-Oliver. Presentación de un caso de expresión incompleta
Adams-Oliver Syndrome: Presentation of an Incompletely Expressed Case
Lourdes González Burgos(1) , Beatriz Di Martino Ortiz(2), Luz Lacarrubba (3).
1. Jefe de Residentes de Dermatología. Cátedra de Dermatología. Hospital de Clínicas. Facultad de Ciencias Médicas. Universidad Nacional de Asunción. Paraguay.
1. Dermatopatólogo. Cátedra de Dermatología. Hospital de Clínicas. Facultad de Ciencias Médicas. Universidad Nacional de Asunción. Paraguay.
2. Dermatólogo. Unidad de Dermatología. Cátedra de Pediatría. Centro Materno-Infantil del Hospital de Clínicas. Universidad Nacional. San Lorenzo. Paraguay.
Correspondencia: Dra. Beatriz Di Martino Ortiz. Paraguarí 1033 casi Teniente Fariña. C.P.: 1325. Asunción-Paraguay.Tel y Fax: (595 21) 446 991. E-mail: beatrizdimartino@gmail.com
Recibido: 12/12/2011, aceptado para publicación: 22/12/2011.
RESUMEN
El Síndrome de Adams-Oliver (SAO) es una genodermatosis infrecuente, con casos esporádicos y familiares, de herencia autosómica dominante, aunque la herencia autosómica recesiva también se ha demostrado, con expresividad variable intra e interfamiliar, caracterizada por la asociación de aplasia cutis congénita en cuero cabelludo, anomalías transversales de los miembros y cutis marmorata telangiectásico congénito. Presentamos el caso de un varón con SAO esporádico, de expresión incompleta, en el que se presentaron aplasia congénita cutis en el cuero cabelludo, cutis marmorata telangiectásico congénito generalizado, criptorquidia y alteración renal. El enfoque terapéutico en este caso fue multidisciplinario.
Palabras clave: Síndrome de Adams-Oliver, aplasia cutis congénita, malformación, genodermatosis.
ABSTRACT
Adams-Oliver syndrome (AOS) is an uncommon genodermatosis with sporadic familial cases that is of autosomal dominant inheritance although autosomal recessive inheritance has also been demonstrated. It is of variable intra- and inter-familial expression and is characterized by the association of aplasia cutis congenita of the scalp, transverse limb anomalies, and cutis marmorata telangiectatica congenita. We present the case of a male patient with sporadic incompletely expressed AOS including aplasia cutis congenita of the scalp, generalized cutis marmorata telangiectatica congenita, cryptorchidism, and renal impairment. The therapeutic focus in this case was multidisciplinary.
Keywords: Adams-Oliver syndrome, aplasia cutis congenita, malformation, genodermatosis.
INTRODUCCIÓN
El síndrome de Adams Oliver es una genodermatosis que se caracteriza por defectos congénitos que afectan el cuero cabelludo, el sistema vascular y las extremidades. Los síntomas varían mucho de paciente a paciente y pueden incluir áreas de piel faltante (con frecuencia en el cuero cabelludo), defectos en las extremidades, defectos cardiacos, cutis marmorata telangiectático congénito, hipertensión pulmonar y alteraciones del sistema nervioso central.
Descrito por primera vez en 1945 por Adams y Oliver quienes reportaron asociación de aplasia cutis congénita y anomalías transversales de los miembros en tres generaciones de una familia(1). De herencia autosómica dominante con expresividad variable intra e inter familiar, recientemente asociada con la mutación del gen ARHGAP31, resultando en pérdida de la actividad del Cdc42 con disrupción de la estructura de actina del citoesqueleto celular(2). También ha sido demostrada la herencia autosómica recesiva(3,4); así como casos esporádicos(3).
Se estima una frecuencia de 0.44/100.000 recién nacidos vivos (o 1 por cada 227.035 recién nacidos vivos). En algunas series se observa con más frecuencia en el sexo femenino (5).
Existen poco más de 100 publicaciones de este síndrome en la literatura médica. Comunicamos el caso de este lactante con la forma esporádica de la enfermedad y expresión clínica incompleta, lo que demuestra el amplio espectro de manifestaciones del síndrome de Adams-Oliver.
CASO CLÍNICO
Lactante varón de 2 meses de edad, nacido por parto vaginal, de embarazo a término y peso adecuado para la edad gestacional. Desde el nacimiento se observan manchas de aspecto reticulado en todo el cuerpo que se acentúan con el llanto y el frío. El cuadro se acompaña de lesión alopécica en cuero cabelludo. Padres no consanguíneos, madre de 47 años, primípara, aparentemente sana y padre de 49 años, aparentemente sano.
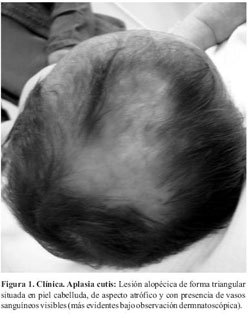
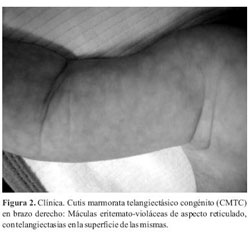
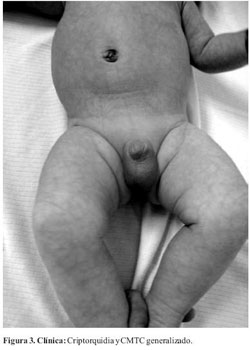
Al examen físico presenta placa alopécica de forma triangular, atrófica, superficie irregular con vasos sanguíneos visibles localizada en vértex (cuero cabelludo) (Figura 1). Se constatan además máculas eritemato-violáceas de aspecto reticulado, con telangiectasias en la superficie de las mismas, de distribución generalizada (Figura 2). Ausencia de testículo en bolsa escrotal derecha (Figura 3). Mancha mongólica en región lumbosacra.
Auxiliares del diagnóstico:
- Hemograma, perfil renal, perfil hepático, eritrosedimentación y PCR dentro de límites normales. VDRL: no reactiva.
- Ecografía testicular: testículo derecho se encuentra en la región inguinal derecha. Ambos testículos presentan características normales.
- Radiografías de miembros superiores e inferiores, ecocardiografía y ecografía transfontanelar normales.
- Ecografía abdominal: ectasia renal izquierda, a controlar.
Anatomía patológica de piel de miembro inferior y cuero cabelludo confirma los diagnósticos de cutis marmorata telangiectásico y aplasia cutis, descartandose otras anomalías.
DIAGNÓSTICO: SÍNDROME DE ADAMS-OLIVER.
DISCUSIÓN
Se han reportado casos esporádicos, sin embargo en la mayoría este síndrome se hereda de forma autosómica dominante.
Es conocida la relación entre la edad avanzada de los padres y la frecuencia de aparición de mutaciones nuevas(5). En el caso de nuestro paciente la madre contaba con 47 años y el padre con 49, por lo que ante la ausencia de antecedentes familiares lo consideramos como un caso esporádico.
Aunque la patogenia es desconocida, se han sugerido mecanismos como predisposición a la secuencia de rotura amniótica, compresión extrínseca, predisposición intrínseca para interferir con el desarrollo del tejido y anomalías vasculares, siendo esta última es la más aceptada(6). Tanto las manifestaciones mayores del SAO, como la presencia de cutis marmorata telangiectásico congénito, dilatación de venas del cuero cabelludo y otras anomalías en los vasos, avalan la “teoría vascular”.
La expresión clínica es variable encontrándose fenotipos de lo más variados. Los hallazgos que se pueden encontrar con más frecuencia son: defectos distales y transversales de las extremidades (84%), aplasia cutis congénita (75%), manifestaciones cardiovasculares (15-20%), cutis marmorata telangiectásico congénito y la dilatación y tortuosidad de las venas del cuero cabelludo (12-20%)(5,7,8). Otras manifestaciones menos frecuentes incluyen, tetillas supernumerarias, criptorquidia, microftalmia, pelo lanoso, paladar hendido, alteraciones renales, hemihipoplasia facial, hipertelorismo, epicantus, hepatoesclerosis, bandas de constricción, malformación arteriovenosa, malformaciones pulmonares, malformaciones del sistema nervioso central(5,7-9). Este paciente presentaba al momento de la consulta con aplasia congénita cutis, cutis marmorata telangiectásico congénito (CMCT), criptorquidia y alteración renal. No se constataron defectos transversales de los miembros por lo que concluimos que se trata de un Síndrome de Adams Oliver de expresión incompleta.
En cuanto al tratamiento que se puede ofrecer a estos pacientes no existe consenso. Este dependerá de los órganos afectos y del grado de compromiso en cada caso; así por ejemplo, en lo que a afección cutánea se refiere el tratamiento varía desde el cierre por segunda intención de la aplasias cutáneas hasta la utilización de injertos de piel y en el caso del CMCT desde el control clínico hasta la realización de LASER vascular(10). En lo que sí existe unanimidad es en la necesidad de manejo multidisciplinario que incluya el consejo genético a los padres y al paciente.
Al hablar de pronóstico también se hace referencia al grado de compromiso de órganos, esto determinará la calidad de vida y la sobrevida de los pacientes con SAO(4,11). El pronóstico en nuestro caso es bueno ya que el paciente contaba con un desarrollo psicomotor aparentemente normal y no presentaba compromiso importante de órganos vitales.
En conclusión podemos decir que la gran heterogeneidad clínica de los pacientes afectados por SAO ha dificultado su consejo genético a los portadores de la anomalía, por lo que representa un reto a la medicina moderna que requiere para su adecuada valoración, prevención, manejo y control en un entorno multidisciplinario.
El presente trabajo tiene como objetivo difundir un caso, de una rara genodermatosis, en la literatura paraguaya con un enfoque multidisciplinario, interrelacionándolo con las distintas áreas médicas involucradas en su tratamiento, destacando la importancia de la dermatología en dicho proceso.
REFERENCIAS
1. Adams FH, Oliver CP. Hereditary deformities in man due to arrested development. J Hered. 1945;39: 3-7. [ Links ]
2. Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, et-al. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet. 2011;88(5): 574-85. [ Links ]
3. Sybert VP. Congenital scalp defects with distal limb anomalies (Adams-Oliver syndrome-McKusik 10030): further suggestion of autosomal recessive inheritance (Letter to the editor). Am J Med Genet. 1989;32:266-67. [ Links ]
4. Klinger G, Merlob P. Adams-Oliver syndrome: autosomal recessive inheritance and new phenotyic-antropometric findings. Am J Med Genet. 1998;79:197-79. [ Links ]
5. Martínez-Frías ML, Arroyo Carrera I, Jiménez Muñoz-Delgado N, Nieto Conde C, Rodriguez Pinilla E, Urioste Azcorra M, et-al. Síndrome de Adams-Oliver en nuestro medio: Aspectos epidemiológicos. An Esp Pediatr. 1996;45:57-61. [ Links ]
6. Patel MS, Taylor GP, Bharya S, AL-Sanna´a N, Adatia I, Chitayat D, et-al. Abnormal pericyte recruitment as a cause for pulmonary hypertension in Adams- Oliver syndrome. Am J Med Genet. 2004;129:294-99. [ Links ]
7. Larralde M, Abad M, Sojo M. ¿Qué síndrome es?. Dermatol Pediatr Lat. 2007;5(2):133-35. [ Links ]
8. Narang T, Kanwar A, Dogra S. Adams–Oliver syndrome: a sporadic occurrence with minimal disease expression. Ped Dermatol. 2008;25(1):115-16.
9. Seo J, Kang J. Lee H, Lee D, Sung H, Hwang S. A Case of Adams-Oliver Syndrome. Ann Dermatol. 2010;22(1):96-98. [ Links ]
10. Llorca M, González de Dios J, Navarro Belmonte MR. Tratamiento con láser de las anomalías vasculares cutáneas en la infancia: análisis prospectivo de nuestra experiencia en 95 niños. An Pediatr. 2000;52:6-14. [ Links ]
11. Bayou F, Boussofara L, Bennani ZL, Ghariani N, Saïdi W, Belajouza C, Denguezli M, Nouira R. Adams-Oliver syndrome. Ann Dermatol Venereol. 2011;138(10):712-14. [ Links ]

