










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPediatría (Asunción)
versão On-line ISSN 1683-9803
Pediatr. (Asunción) v.37 n.1 Asunción abr. 2010
CASO CLÍNICO
Enfermedad de Moya-Moya
Moya-Moya Desease
Ángel R. Acuña F.(1), Cristina O. de Godoy (2).
1. Ex Jefe de Servicio. 2. Jefe de Sala. Servicio de Pediatría. Hospital Rigoberto Caballero. Asunción-Paraguay.
Solicitud de sobretiros: Dr. Ángel R. Acuña F. Arecutacuá 2260. San Vicente. Asunción-Paraguay. E-mail: aracunaf@hotmail.com
Articulo recibo en marzo de 2010, aceptado para publicación abril 2010.
RESUMEN
Enfermedad esteno-oclusiva de la porción terminal de las arterias carótidas internas y de sus ramas principales (arteria carótida media y arteria carótida anterior), progresiva, prevalente en el este Asiático (Japón, Corea), muy rara en nuestro país como en el resto del mundo, de causa desconocida. Se manifiesta mas frecuentemente como un accidente cerebrovascular isquémico transitorio o permanente en los niños menores de 10 años y como accidente cerebrovascular hemorrágico en el adulto. El diagnóstico se realiza con la Angiografía Digital Cerebral y el tratamiento consiste en la revascularización cerebral, que mejora el cuadro neurológico y la calidad de vida.`Se presenta el caso de una niña de 3 años de edad con convulsiones, afasia de expresión, lúcida y hemiparesia izquierda, que luego de una franca mejoría vuelve a presentar una semana después, otra crisis más severa con compromiso de conciencia y hemiplejía derecha, cuyo estudio neurorradiológico reveló la imagen típica de volutas de humo de cigarrillo en la base del cerebro, característico de la enfermedad de Moya-Moya e intervenida quirúrgicamente con la revascularización cerebral por medio de la técnica: EncefaloDuroArterioSinangiosis (EDAS), con buen resultado.
Palabras Claves: Enfermedad de Moya- Moya, accidente cerebrovascular, angiografía digital, revascularización cerebral
ABSTRACT
Moya-Moya is a progressive steno-occlusive disease of the internal carotid arteries and its major branches (middle cerebral artery, and anterior cerebral artery) whose cause is unknown, and which is reported primarily in east Asia (Japan and Korea) but rare in Paraguay and the rest of the world. In children under age 10, it presents most commonly as a transient or permanent ischemic attack and in adults as a hemorrhagic stroke. Diagnosis is done by digital subtraction cerebral angiography, and treatment consists of cerebral revascularization to improve the neurological profile and quality of life. We present the case of a 3-year old female child with seizures and motor aphasia who was lucid, with left hemiparesis, and who after complete recovery presented again a week later with a more severe attack, with impaired consciousness and right hemiplegia. Radiological study revealed a typical image of swirls of 'cigarette smoke' at the base of the brain that are characteristic of Moya-Moya disease, and who was operated on using encephaloduroarteriosynangiosis (EDAS), with good results.
Keywords: Moya-Moya disease, cerebrovascular accident, cerebral revascularization, digital subtraction angiography.
INTRODUCCIÓN
La Enfermedad de Moya-Moya es una patología neurológica de la infancia, de evolución crónica, de presentación infrecuente, caracterizada por la estenosis u oclusión bilateral, progresiva de la porción supraselar de la arteria carótida interna (ACI) y sus ramas principales, arteria carótida media (ACM) y arteria carótida anterior (ACA) y la formación de circulación colateral en la base del cerebro capaces de provocar signos y síntomas compatibles con accidentes cerebro vasculares de tipo isquémico transitorio o permanente. Descripta por primera vez hace poco más de 50 años en dos niños japoneses y cuyo nombre se debe a la imagen digital observada en la base del cerebro ocasionado por dicha circulación colateral, que asemeja a las volutas del humo de cigarrillo, y se expresa como Moya-Moya en lengua japonesa (1-3).
Se presenta con mayor frecuencia en la edad pediátrica, sobre todo en niños menores de 10 años(3), y menos frecuentemente en adultos hacia la tercera y cuarta décadas de la vida (4-5). Se describe un franco predominio en el sexo femenino (1,3,6,7), se reporta ampliamente la alta prevalencia en poblaciones del este asiático, sobre todo Japón y Corea, y mas raramente en otras partes del mundo (4,6-8).
La etiología de la enfermedad sigue siendo desconocida, pero se han publicado las implicancias genéticas y ambientales en el desarrollo de algunos casos, específicamente se le ha relacionado con alteraciones de los cromosomas 3, 6 y 17 (7,9), la incidencia de un 10% de casos familiares y la alta prevalencia en japoneses indican fuertemente la presencia de factores genéticos (5). Se sospecha que factores ambientales asociados a factores genéticos, como la radioterapia en procesos intracraneanos que incrementan el riesgo de aparición de Moya-Moya, siendo un ejemplo clásico la neurofibromatosis tipo 1(8,9). Paralelamente se describe el desarrollo de estenosis progresiva de la parte terminal de la ACI y sus ramas principales y formación de vasos colaterales tipo Moya-Moya en algunas enfermedades congénitas y adquiridas como: síndrome de Down, esclerosis tuberosa, neurofibromatosis tipo 1, anemia a célula falciforme, algunas infecciones de cabeza y cuello, trauma craneano, irradiación cerebral, cardiopatías congénitas (3,5,8,10). Además la literatura reporta la persistencia de arterias embrionarias como la trigeminal y más raramente la arteria de Bernasconi-Cassinari que plantearía un inicio intrauterino de la enfermedad, desde el mismo momento del desarrollo arterial en la etapa embrionaria por predisposición constitucional genética (11).
Clínicamente el cuadro se presenta como un accidente cerebrovascular isquémico transitorio o permanente o cambiante, con hemiplejía o hemiparesia, convulsiones, trastornos del lenguaje (disartria o afasia), epilepsia y otros síntomas atípicos como: dificultad del aprendizaje, comportamiento anormal, caídas bruscas, cefaleas agudas o crónicas. La otra forma de presentación es la de un accidente cerebrovascular hemorrágico, y es mas común en la tercera y cuarta décadas de la vida (1,5,11).
El procedimiento diagnóstico inicial (6,8,9) consiste en la realización de una tomografía axial computarizada (TAC) y/o resonancia magnética nuclear (RMN) del cráneo donde se evidencia el ACV tipo isquémico o hemorrágico, seguidas de angiorresonancia magnética nuclear (ARMN) y/o angiografía cerebral de los 4 vasos o troncos con sustracción digital (AD), siendo éste ultimo procedimiento neurorradiológico el gold standard diagnóstico(3), donde se observan los cambios esteno-oclusivos de la ACI bilateral y de sus ramas principales y la imagen típica de volutas de humo de cigarrillo dadas por los vasos colaterales que se forman alrededor del polígono de Willis en la base del cerebro, y da el sello diagnóstico de la enfermedad (1,5,9,10,11).
Suzuki y Takaku (1) en 1969 describieron 6 etapas evolutivas de la esteno-oclusión vascular a través de la imagen angiográfica:
1. Estrechamiento de la bifurcación de la carótida sin otras anomalías
2. Iniciación de Moya-Moya: Las arterias intracerebrales principales están dilatadas. La forma de Moya-Moya es ligeramente observable. No existe circulación colateral demostrada de la arteria cerebral externa hacia el área intracraneal.
3. Intensificación de Moya-Moya: El fenómeno Moya-Moya es claramente demostrable. Los cambios aparecen en las arterias cerebrales principales, anterior y media, éstos cambios se ven en la periferia de la ACA, la mitad de la ACM está perdida y reemplazada por el Moya-Moya, observada como una vascularización en forma de nube.
4. Minimización de Moya-Moya: La oclusión de la ACI se extiende hasta su unión con la comunicante posterior, la arteria carótida posterior, la cual era visualizada; en ésta etapa desaparece del angiograma y los vasos Moya-Moya se adelgazan y forman una red pobre con poco rendimiento. Hay aumento de la circulación extracraneal.
5. Reducción de Moya-Moya: En ésta etapa todas las arterias principales que salen de la ACI desaparecen por completo. La tendencia de la reducción que se ha iniciado en la etapa anterior va progresando, el Moya-Moya se hace más pobre y limitado al sifón carotídeo. La circulación colateral de la ACE continúa incrementándose.
6. Desaparición de Moya-Moya: En ésta etapa el sifón de la ACI desaparece completamente, también está perdida la típica imagen de Moya-Moya en la base del cerebro y solo se visualiza la circulación colateral de la ACE en la entrada del área intracraneal. En ésta fase terminal de la enfermedad la circulación cerebral es mantenida por la ruta de la ACE o de la arteria vertebral.
El Comité de Investigación de la oclusión espontánea del Circulo de Willis del Ministerio de Salud y Bienestar del Japón definió los criterios diagnósticos de la Enfermedad de Moya-Moya: (1978)(2,3,5).
1. Cambios esteno-oclusivos en la porción terminal de la ACI y sus principales ramas y desarrollo de vasos Moya-Moya en la base del cráneo.
2. Los cambios deben ser bilaterales.
3. Causa desconocida.
Sin embargo ésta condición patológica puede ser observada en el contexto de otras enfermedades congénitas o adquiridas, llamándose a éstos Síndrome o Cuasi Moya-Moya.
Hay casos en que los cambios permanecen invariables en su localización unilateral y sin causa demostrable, a éstos se les llama Enfermedad de Moya-Moya unilateral.
En un estudio electroencefalográfico hecho en Seúl (Corea) en 22 casos seleccionados de una serie mayor de pacientes que cumplieron con el criterio de inclusión, (realización de EEG e hiperventilación), en el pre y post-operatorio de la enfermedad de Moya-Moya sometido a revascularización indirecta, el hallazgo consistió en ondas lentas de gran amplitud, llamadas enlentecimiento hemisférico posterior o enlentecimiento centrotemporal, que a menudo aparecen relacionadas a isquemia cerebral, y lo característico de la enfermedad de Moya-Moya, el “fenómeno de reconstrucción”(re-build up), que se caracteriza por la aparición de ondas monorrítmicas generalizadas y ondas lentas de alto voltaje durante la hiperventilación (durante 3 min.), seguidas por el fenómeno de la reconstrucción con la reaparición de ondas lentas de gran amplitud varios minutos después (5-7 min.) de finalizada la hiperventilación; posteriormente, el EEG registra nuevamente la actividad basal.
El mecanismo responsable del fenómeno no está bien claro, pero se sugiere que durante la hiperventilación hay una reducción de la presión parcial del dióxido de carbono en las arterias, en cambio la concentración de la hemoglobina oxigenada se incrementa; y consecuentemente lo contrario, con la reducción de la perfusión cerebral 5-7 minutos después de la hiperventilación. El estudio reveló, en el preoperatorio, el fenómeno de reconstrucción en alrededor del 80% de los casos y la desaparición o la reducción y distribución del mismo 6 a 12 meses después de la cirugía y con buena correlación con la mejoría clínica y con la perfusión cerebral en el SPECT (Tomografía por emisión de fotón único).Los autores sugieren realizar el EEG con hiperventilación, que es un estudio simple, no invasivo y aceptado por los niños, como un test primario, útil para el seguimiento, después de la cirugía (6).
Para el tratamiento se ha recurrido a diversos recursos farmacológicos como antiagregantes plaquetarios (aspirina), anticoagulantes (cumarinicos, heparina de bajo peso molecular), vasodilatadores (calcio antagonistas), que no brindaron el resultado esperado (9,11).
Los que sí mostraron mejores resultados en el pronóstico y calidad de vida son los procedimientos quirúrgicos de revascularización cerebral que son de dos tipos: directos e indirectos: la técnica quirúrgica directa o bypass, como la anastomosis de la arteria temporal superficial (ATS) con la arteria cerebral media, que presenta dificultades técnicas por el pequeño calibre de los vasos y las indirectas como: La Encefalo Duro Arterio Sinangiosis (EDAS) que consiste en la liberación de la ATS y unión al borde de la incisión de la duramadre, técnica modificada por su propio creador, en la que se realiza una apertura mas amplia de la duramadre, con aposición de la ATS y amplio tejido galeal circundante y fijación de los bordes de éste tejido a la piamadre de la convexidad cerebral a la que se efectúan pequeñas ventanas, permitiendo un contacto mas estrecho de la arteria donante con el parénquima cerebral, técnica utilizada por Quintana Marin con resultado excelente en 9 de 10 casos(5). La Encefalo Mio Sinangiosis (EMS) en que se realiza la aposición del músculo temporal sobre la superficie del cerebro y así inducir el desarrollo de vaso colaterales; y la Encefalo Duro Arterio Mio Sinangiosis (EDAMS) que es una técnica combinada de las dos anteriores (2,7,9,11).
El pronóstico no es tan alentador en los pacientes menores de dos años, el mejor resultado se observa en los niños mayores si la detección de la oclusión arterial se hace precozmente, sobre todo antes de desarrollar un infarto cerebral o hemorragia, y si se realiza la cirugía de revascularización, cuyo resultado produce una mejor calidad de vida, a pesar de que en el seguimiento por estudio arteriográfico se constate la progresividad esteno-oclusiva de los vasos involucrados (7).
CASO CLÍNICO
K.R.F. preescolar de 3 años de edad de sexo femenino, producto de una primera gestación no complicada de padres sanos, no consanguíneos, nacida de un parto normal sin complicaciones, con crecimiento y desarrollo normal. Ingresa al Hospital Rigoberto Caballero (22.05.07), derivada del Hospital Santa Rosa de Lima de Cnel. Oviedo por un cuadro de 24 hs de evolución de convulsiones tipo tónicoclónicas del hemicuerpo izquierdo, sin fiebre, quedando luego con paresia faciobraquiocrural izquierda, afasia de expresión, con dificultad para la marcha y trastornos en la deglución, lúcida y comprende consignas verbales, PA: 100/60. Se practica TAC de cráneo sin contraste (25.05.07), que revela hipodensidad encefálica cortical temporal derecha y compresión parcial del ventrículo ipsilateral, que sugiere área de isquemia cortical temporal derecha. Se la trata con difenilhidantoina (DFH) y dexametasona.
Se le practican los siguientes estudios laboratoriales: hemograma, eritrosedimentación, PCR en varias oportunidades que fueron normales. Frotis de sangre periférica y orina normales. Serología para Herpes Virus I y II IgG e IgM y búsqueda de antígenos de virus respiratorios: (-) negativos. Urea: 27 mg%, Creatinina: 0.4mg%, GOT: 48us/L, Fosfatasa Alcalina: 212us/L, Bilirrubina Total: 0.2mg%, Colesterol total: 194mg%, Triglicéridos: 63mg%, Acido Úrico: 2.6mg%, Glicemia: 154mg%, que se repite: 93mg%. Artritest (FR), ANA y AntiDNA (-) negativos. VDRL: (-). Anticoagulante Lúpico (AL): (+) 80 a 120% de actividad.
El Ecocardiograma/Doppler: resultó normal. Se realiza Angiotomografía computarizada multislice de cráneo con contraste (26.05.07), que muestra imágenes sugerentes de alteración obstructiva en el territorio de la arteria Silviana derecha en su rama externa temporoparietal y estrechamiento de la ACI derecha y en menor grado en el lado izquierdo, con vascularización aumentada de las Tálamo-perforantes, que sugieren una probable estenosis progresiva de la misma. En el territorio posterior, ambas arterias Vertebrales, Basilar y Cerebrales posteriores exhiben densidad y trayecto habitual. Los datos son compatibles con Enfermedad de Moya-Moya y se plantea la realización de una Angiografía Digital (AD).
En espera de dicho estudio la paciente muestras signos de mejoría del déficit motor izquierdo y se reciben los estudios de procoagulabilidad: Proteína C: 103% de actividad (80 a 120%), Proteína S: 120% de actividad (80 a 120%), Antitrombina III: 120% de actividad (70 a 120%), Factor V de Leyden: resistencia normal a la proteína C activada, Homocisteína: 5.6 umol/L ( 5 a 12 umol/L) y un coagulograma:TP: 12” 100%, TTPA: 26.20” (26.1-36.3”), Fibrinógeno: 200 mg% (200-400 mg%), Tpo. Coag.: 6´min., Tpo. Sangría: 1´min., Plaquetas: 550.000, repetido: 356.000.
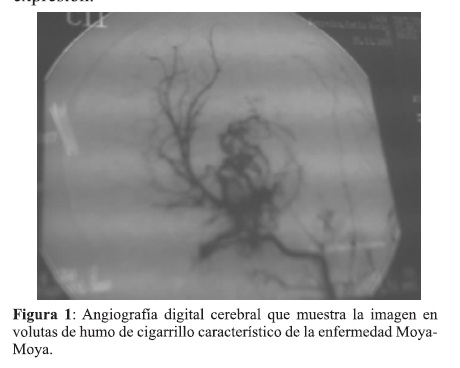
Estando la niña en franca mejoría, bruscamente vuelve (31.05.07) a presentar un nuevo episodio de crisis convulsiva generalizada con carácter de status , parálisis faciobraquiocrural derecha y compromiso progresivo de la conciencia lo que motiva su ingreso a la unidad de cuidado intensivo por 12 hs., se le instala anticoagulación con heparina de bajo peso molecular (HBPM) y sigue con DFH. De regreso a sala, se le suspende HBPM, sin convulsiones, recupera la conciencia, no así el compromiso motor; el 4.06.07 se le practica Angiografía cerebral de 4 troncos con Radiología Digital y Sustracción electrónica de imágenes (AD) (Figura 1), que muestra importante estrechamiento de las porciones terminales de ambas ACI, con derivación del flujo sanguíneo hacia las Arterias oftálmicas y recanalización hacia las arterias ramas, Silviana y Cerebral anterior; notándose el mayor flujo por el lado izquierdo que el derecho. Se observa buena perfusión en las Vertebrales, Basilar y Comunicante Posterior, visualizándose importante desarrollo de ramas colaterales, Tálamo Perforante o Tálamo Estriadas, en forma bilateral y múltiple, principalmente en el lado izquierdo, dando la imagen parecida a las volutas del humo de cigarrillo, característico de la Enfermedad de Moya-Moya, que confirma el diagnóstico de dicha enfermedad y se indica el tratamiento neuroquirúrgico de revascularización; para tal efecto es derivada al Centro Materno Infantil (Hospital de Clínicas), de alta complejidad, donde se realiza dicho procedimiento (20.06.07), por medio de la técnica EncefaloDuroArterioSinangiosis, empleando a la arteria carótida externa rama temporal superficial, como arteria donante. La intervención trascurrió sin inconvenientes, la niña fue recuperando progresivamente sus funciones motoras, persistiendo una moderada afasia de expresión.
En el seguimiento por control ambulatorio 2 meses despu és, se constató diplejía espástica bilateral a predominio derecho y en la AD se advierte prolongación del tiempo vascular parenquimatoso que muestra mejor distribución córtico-subcortical a predominancia izquierda. Irrigación en territorio posterior no modificado, en relación a estudios anteriores. La opinión neuroquirúrgica define que no es necesaria una cirugía del lado izquierdo, por el momento. Al año del post-operatorio, la paciente acude al control caminando, con minima afasia de expresión y una discreta espasticidad de manos.
DISCUSIÓN
La Enfermedad de Moya-Moya es una patología extremadamente rara en nuestro país. En la Cátedra de Pediatría de la FCM-UNA (Hospital de Clínicas), centro de referencia importante del país, no existe registro de ésta enfermedad. Sin embargo en una publicación nacional(12), se presenta como una variable estadística diagnóstica, 10 pacientes de 2 a 15 años de edad, un caso de Síndrome de Moya-Moya like, que se presento como un ACV hemorrágico intraventricular y diagnosticado por Angiografía Digital, con imagen en pico de flauta, de causa desconocida, y no refieren la coexistencia de alguna enfermedad subyacente o asociada, por lo que consideramos que el presente caso es el primero diagnosticado en el país, que se ajusta a los criterios diagnósticos del Ministerio de Salud y Bienestar del Japón, descrita mas arriba.
La bibliografía reporta ampliamente la rareza de la enfermedad fuera del este Asiático (Japón, Corea) donde la prevalencia es 10 veces mayor de 3.16 casos por 100.000 habitantes/año(4-7). La edad y el sexo de nuestra paciente se corresponde con lo descrito en la literatura(1-3,5-8,11).
La presentación clínica en el caso de esta paciente fue de presentación aguda, con convulsiones generalizadas, déficit motor en un hemicuerpo, con mejoría posterior y luego otra crisis con focalización en el otro hemicuerpo, con compromiso del lenguaje y del sensorio, que representa la forma mas frecuente, describiéndose además, hemiplejías a repetición, crisis convulsivas epileptiformes y otros síntomas menores(2,3,8,9,11).
A su ingreso hospitalario y mientras se tomaban las medidas de sostén y control de los síntomas, se realizó TAC de cráneo, que reveló una hipodensidad encefálica cortical temporal derecha sugerente del diagnóstico de ACV isquémico, con lo cual se inician las pesquisas correspondientes para determinar el probable responsable etiológico que pudiera estar involucrado, en espera de la asistencia del neurólogo infantil, siguiendo la siguiente línea de investigación(13).
1. Traumatismo de faringe (caída con un objeto punzante en la boca del niño, lesión arterial, formación de trombo y siembra de émbolos).
2. Infección de cabeza, cuello, absceso retrofaríngeo, varicela, herpes virus y otras.
3. Enfermedades de la sangre como: anemia falciforme, leucemia, Trombofilias por déficit de proteína C, proteína S, antitrombina III, factor V de Leyden, mutación de la protrombina (G20210A) y síndrome antifosfolípido.
4. Cardiopatías congénitas.
5. Trastorno metabólico: homocistinuria.
Todas estas patologías fueron descartadas por la clínica y los estudios laboratoriales y de imágenes, que retornaron normales, salvo el anticoagulante lúpico (AL), que fue (+), lo que fue considerado como un hallazgo casual, ya que así se describe como consecuencia de infecciones o vacunaciones previas, de tipo transitorio y no ocasionan trombosis(14). Al 5º día de internación se realiza ARMN con datos sugerentes de estrechamiento de la ACI y de la Silviana derecha, que hacen sospechar enfermedad de Moya-Moya, que se confirma con Angiografía por sustracción Digital hecha 9 días después, que evidencia importante estrechamiento de las porciones terminales de ambas ACI y derivación del flujo hacia las Arterias Oftálmicas, con importante desarrollo de vasos colaterales del polígono de Willis, dando una imagen típica de volutas de humo de cigarrillo en la base del cráneo; momento en que además se excluyeron otras patologías sistémicas con clínica neurológica y cambios vasculares similares, como: síndrome de Down, neurofibromatosis tipo 1, esclerosis tuberosa, enfermedad de Beçet, displasia fibromuscular, enfermedad de Hirschprung, síndrome de Marfán, síndrome de Apert, antecedentes de radioterapia cerebral, acorde con lo que se describe en la literatural mundial(1-3,5,9), por lo que puede definirse el caso estudiado, como Enfermedad de Moya-Moya, en el que no se logró demostrar la causa.
Si bien es cierto que en la búsqueda de la causa del padecimiento de un paciente, es necesario orientar la misma en razón a la frecuencia de la patología regional, ello no debe implicar el desdeño de las rarezas, y por lo tanto siempre se impone profundizar la investigación y recurrir a los estudios que permitan develarlas, para ofrecer el tratamiento precoz y adecuado, como ocurrió en el caso que se publica, que permitió la realización de la única opción terapéutica, la revascularización cerebral y con buen resultado, como beneficio inobjetable para en principio superar el riesgo vital y luego asegurar calidad de vida.
REFERENCIAS
1. Suzuki J, Takaku A. Cerebrovascular “Moya-Moya” Disease. Disease Showing Abnormal Net-Like Vessels in Base of Brain. Arch Neurol. 1969;20:288-299. [ Links ]
2. Galicchio S, Maza E, Jaimovich R, Arroyo H A. Enfermedad de Moya- Moya. Arch Arg Pediatr. 1998;96:263-267. [ Links ]
3. Urrutia Ruiz M, Barragán Pérez E, Hernández Aguilar J, Garza Morales S, Cruz Martínez E, Santana García F, et-al. Bol Med Hosp Infant Mex. 2007;64:99-105. [ Links ]
4. Marcikevicius E, Liutkus D, Gvazdaitis A. Experience of treatment of Moya-Moya disease at the clinic of neurosurgery of Kaunas University of Medicine. Medicina (Kaunas). 2006;42(2):130-136. [ Links ]
5. Quintana Marin L. Experiencia de 20 años en el manejo de la Enfermedad de Moya-Moya. Rev Chil Neuroc. 2004;23:30-36. [ Links ]
6. Kim D S, Ko Y S, Choi C G. Postoperative Electroencephalogram for Follow up of Pediatric Moya-Moya Disease. J Korean Med Sci. 2006;21:495-499. [ Links ]
7. Ajler P M, Brocanelli M A, Ajler G S. Enfermedad de Moya-Moya: reporte de un caso. Rev Arg Neuroc. 2005;19:146-148. [ Links ]
8. González G, Russi M E, Campistol J, Costa G, Navarro Balbuena R, Crosa R, et-al. Moya-Moya en la población infantil no asiática: ampliación de casuística. Arch Pediatr Urug. 2008;79(4):291-302. [ Links ]
9. Yañez P L, Paredes M M, Lapadula A M, Martínez T P, Duran H F. Enfermedad de Moya-Moya: a propósito de dos casos. Rev Chil Pediatr. 2008;79(6):629-635. [ Links ]
10. Cornelio Nieto J O, Dávila Gutiérrez G, Ferreiro Irigoyen R, Alcalá H. Hemiplejia aguda de la infancia y hemiconvulsiones alternantes secundarias a enfermedad de Moya-Moya: reporte de 1 caso asociado a síndrome de Down. Bol Hosp Infant Mex. 1990;47(1):39-42. [ Links ]
11. Castroviejo IP, Pascual Pascual SI, Velazquez R, Viaño J, Martínez V. Síndrome de Moya-Moya: seguimiento de 12 pacientes. Neurología. 2006;21(10):695-703. [ Links ]
12. Nunes T, Recalde L, Espínola de Canata M. Características de los accidentes cerebrovasculares (ACV) en niños. Pediatr (Asunción). 2008;35(1):18-23. [ Links ]
13. Johnston M V. Síndrome vasculares cerebrales agudos. En: Behrman R E, Kliegman R M, Jenson H B. editores. Nelson Trat.de Pediatría. 17 edición. España: Elsevier; 2004.p.2035-2038. [ Links ]
14. Ravelli A, Martini A. Síndrome antifosfolípido. Clin Pediatr Norteam. 2005;52(2):469-491. [ Links ]

