










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPediatría (Asunción)
versão On-line ISSN 1683-9803
Pediatr. (Asunción) v.36 n.3 Asunción dez. 2009
CASO CLÍNICO
Monosomía Parcial del Brazo Corto del Cromosoma 9.
Partial Monosomy of the Short Arm of Chromosome 9
Torres E1, Monjagata N1, Herreros MB1,2, Ascurra M3, Rodríguez S1, Ayala A1.
1. Docentes investigadoras del Departamento de Biología Molecular y Genética. Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción.
2. Medica Genetista del Instituto de Protección a personas con capacidades especiales - INPRO.
3. Docente investigadora del Departamento de Biomedicina. Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción.
Solicitud de Sobretiros: Lic. Elodia Torres. Instituto de Investigaciones en Ciencias de la Salud. Río de la Plata y Lagerenza. E-mail: genetica@iics.una.py, elo_torres@yahoo.com.ar
Artículo recibido el 30 de Setiembre de 2009, aceptado para publicación 26 de Noviembre de 2009.
RESUMEN
Existen aproximadamente 100 casos reportados con monosomia parcial del brazo corto del cromosoma 9 a nivel mundial. Se trata de una aberración cromosómica estructural rara donde aproximadamente el 85% de los casos, la delección corresponde a una mutación de novo, esporádica y espontánea. Ocurre muy tempranamente durante el desarrollo embrionario, por razones aún desconocidas y por lo general involucra a la porción del cromosoma 9p22. Esta cromosomopatía podría sospecharse desde el nacimiento, por las características fenotípicas faciales y la presencia de pliegues palmares profundos. Se describe el caso de una paciente de sexo femenino de 15 años, a quién se le realizó el estudio citogenético por retardo mental y dismorfias varias. El estudio cromosómico llevado a cabo en sangre periférica reveló el siguiente resultado: 46,XX,del(9p22). La realización temprana del estudio citogenético en pacientes portadores de retardo mental y dismorfias es de gran importancia ya que permite establecer el diagnóstico y el pronóstico del paciente y realizar el correspondiente asesoramiento genético familiar.
Palabras claves: Monosomía, cromosoma 9, dimorfismo, retardo mental.
ABSTRACT
Worldwide, approximately 100 cases of partial monosomy of the short arm of chromosome 9 have been reported. The condition is a rare structural anomaly of the chromosome that in approximately 85% of cases represents a deletion due to a mutation that is de novo, sporadic, and spontaneous. The mutation occurs very early in embryonic development for reasons that are yet unknown, and generally involves a portion of the chromosome 9p22. This chromosomal condition may be suspected from birth due to phenotypic facial characteristics and the presence of deep palm creases. We describe a 15-year old female patient on whom a cytogenetic study was done due to mental retardation and various malformations. The chromosomal study, done using peripheral blood, showed 46,XX of the 9p22. Early performance of cytogenetic studies in patients with mental retardation and malformations is important in establishing a diagnosis and prognosis for the patient and for performing the appropriate family genetic counseling.
Key words: Monosomy, chromosome 9, malformation, mental retardation, chromosome disorders.
INTRODUCCIÓN
La monosomía parcial del brazo corto del cromosoma 9 constituye el Síndrome de la deleción 9p, este trastorno constituye una aberración estructural rara y desde su descripción inicial en 1973 por Alfi et al, se han reportado aproximadamente 100 casos (1,2). Los pacientes con esta anomalía presentan en general un crecimiento normal. El coeficiente intelectual (CI) medio es de 49, con un intervalo comprendido entre 33 y 73 y con frecuencia son personas muy sociables. A nivel creaneofacial presentan las siguientes características clínicas: craneosinostosis con afectación de sutura metópica que causa trigonocefalia; occipucio plano. Además presentan; fisuras palpebrales inclinadas hacia arriba, epicantus, ojos prominentes secundarios a crestas supraorbitales hipoplásicas; cejas arqueadas y altas; hipoplasia mediofacial con nariz corta, depresión del puente nasal y filtrum alargado; boca pequeña, micrognatia; orejas malformadas y dismórficas, de implantación baja y rotadas hacia atrás, con lóbulos hipoplásicos y adherentes; cuello corto y ancho con línea de implantación del cabello baja. En las manos, pliegues palmares únicos, las falanges medias de los dedos son cortas con pliegues de flexión extra, falanges distales y uñas cortas, con un exceso de patrones en espiral en las huellas dactilares de las puntas de los dedos, y defectos de posición del pie. A nivel cardiovascular presentan comunicación interventricular; persistencia del conducto arterioso y/o estenosis pulmonar (1-5). Entre otras manifestaciones reportadas se describen: escoliosis, pezones ampliamente espaciados, separación de los músculos rectos del abdomen, hernia inguinal y/o umbilical, micropene y/o criptorquidia en los varones, y labios hipoplásicos en las mujeres (1,2). Entre las anomalías ocasionales se citan: ptosis, paladar hendido, atresia de coanas, polidactilia post axial, hernia diafragmática, hidronefrosis; anomalías radiológicas en costillas, clavículas y vértebras (1,2,6,7).
DESCRIPCIÓN DEL CASO CLÍNICO
Adolescente de 15 años de edad, hija de madre de 54 años y padre de 61 años, al momento de la consulta. Madre sana, padre alcohólico, sin consanguinidad. Sin antecedentes familiares Figura 1, ni antecedentes patológicos durante el embarazo. La niña nace a las 40 semanas de gestación en un parto normal con hipoxia perinatal, peso al nacimiento 2.400 grs. Presentó retardo del desarrollo sicomotor con marcha a los 2 años y 6 meses. Menarca a los 13 años con menstruaciones normales y regulares 30/3-4. 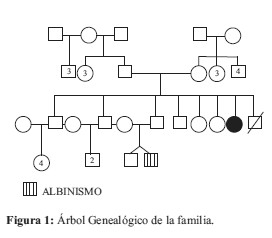
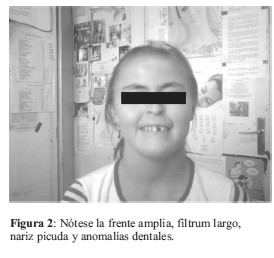
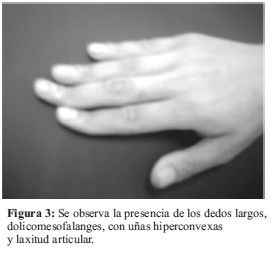

El estudio cromosómico fue llevado a cabo en sangre periférica a través del cultivo de linfocitos (8-10); fueron analizadas 50 metafases con bandas G y C, mediante el software para análisis cromosómico VideoTesT- Kario 3.1, donde se reveló la presencia de una deleción en el brazo corto del cromosoma 9 (Figura 5 y 6). Siendo el cariotipo de la paciente 46,XX,del 9(p22).


DISCUSIÓN
La deleción 9p es conocida por la heterogeneidad y variabilidad en el sitio de ruptura, pero la región crítica corresponde a un intervalo de 4-6 Mb conocida como región 9p22, la misma deleción encontrada en la paciente, que se utiliza como consenso para el delineamiento del fenotipo en estos pacientes (7).
En alrededor del 85% de los casos, la deleción corresponde a una mutación de novo, esporádica y espontánea que ocurre muy tempranamente durante el desarrollo embrionario, producido por razones desconocidas y que por lo general involucra a la porción distal 9p22; el 15% restante resulta de una translocación parental por lo que los progenitores deben ser estudiados (1,4). Los cariotipos de los padres aún no se ha realizado, por lo que se desconoce el origen de la delección; pero en cuanto al riesgo de recurrencia, si el cariotipo de los padres fuera normal se presume que este sería negligible y si uno de los padres fuese portador el riesgo de recurrencia en la familia sería alto (4). Suponemos que este no es el caso de la paciente porque se trata de la séptima hija y no existen antecedentes previos en la familia (Fig. 1).
El diagnóstico de esta enfermedad debería ser realizado en forma precoz, de ser posible al nacimiento (1,4,11). El caso en estudio no se ha beneficiado con ello, debido al bajo nivel sociocultural de la familia, y solo consultaron cuando la joven presentó trastornos siquiátricos, los cuales son comunes en este Síndrome a medida que avanza la edad (12). Esto último resalta la importancia de sugerir la consulta con un genetista ante la presencia de pacientes portadores de retardo mental con malformaciones diversas, a fin de establecer el diagnostico y realizar el asesoramiento genético familiar con respecto a las características, el manejo y la forma de herencia de la patología en cuestión.
REFERENCIAS
1. Gorlin RJ, Cohen MM, Hennekam RCM. Chromosomal syndromes: unusual variants. En: Síndromes of the Head and Neck. 4th ed. London: Oxford University Press; 2001.p.85-5. [ Links ]
2. Jones K. Smith patrones reconocibles de la malformación humana. 6ª ed. Madrid: Elsevier; 2006. [ Links ]
3. Alfi OS, Donnell GN, Allderdice PW, Derencsenvi A. The 9p- syndrome. Ann Genet. 1976;19(1):11-16. [ Links ]
4. Buyse ML. Birth defects encyclopedia: a service of the Center For Birth Defects Information Services. USA: Blackwell Scientific Publications; 1990. [ Links ]
5. Christ LA, Crowe CA, Micale MA, Conrox JM, Schawrtz S. Chromosome breakage hotspots and delineation of the critical region fon de 9p- deletion syndrome. Am J Hum Genet. 1999;65(5):1387-95. [ Links ]
6. ShashiV, Golden WL, Fryburg JS. Choanal atresia in a patient with the deletion (9p) syndrome. Am J Med Genet. 1994;49(1)88-90. [ Links ]
7. Swinkels ME, Simons A, Smeets DF, Vissers LE, Veltman JA. Clinical an cytogenetic characterización of 13 dutch patients with deletion 9p syndrome: delineation of the critical region for a consensus phenotype. Am J Med Genet. 2008;1464(11):1430-38. [ Links ]
8. Seabright M. A rapid banding technique for human chromosome. Lancet. 1971;2:971. [ Links ]
9. Summer A. A simple technique for human centromeric heterochromatin. Exp Cell Res. 1972;75:304-06. [ Links ]
10. Verma R, Babu A. Human chromosomes (manual of basic techniques). USA: Pergamon Press; 1989. [ Links ]
11. Huret J, Leonard C, Forestier B, Tethoré MO, Lejeune J. Eleven new cases of del (9p) and features from 80 cases. J Med Genet. 1988;25(11):741-49. [ Links ]
12. Hou JW. Del (9p) syndrome: report of four cases. Acta Paediatric Taiwan. 2003;44(1):50-53. [ Links ]

