










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPediatría (Asunción)
versão On-line ISSN 1683-9803
Pediatr. (Asunción) v.35 n.2 Asunción 2008
CASO CLINICO
Cromosoma 20 en Anillo en Gemelas Monocigotas
Ring Chromosome 20 in Monozygotic Twins
Ascurra M1, Rodrigue S1, Herreros M2, Torres E1.
1. Laboratório de Genética, Instituto de Investigaciones en Ciencias de la Salud (IICS)
2. Instituto Nacional de Protección a Personas Excepcionales (INPRO).
RESUMEN
Introducción: El cromosoma en anillo es una infrecuente alteración cromosómica caracterizada por la delección del cromosoma en ambos extremos y su posterior ensamblaje en forma circular. El fenotipo y la clínica del paciente se hallan en relación directa con la cantidad de material genético perdido en los extremos así como el cromosoma involucrado, pudiendo incluso esta anomalía estar presente sin repercusión clínica. El síndrome del cromosoma 20 en anillo se caracteriza a su vez, por retraso mental, trastornos de conducta, dismorfias y epilepsia refractaria con crisis polimorfas, siendo el tercer tipo de epilepsia conocido de base genética localizado en el cromosoma 20, en el locus 20q13.
Caso Clínico: Dos lactantes, de 9 meses de edad, que consultan por retraso psicomotor y rasgos dismórficos. Producto de un embarazo gemelar monocorial-monoamniotico, parto por cesárea, ambas ingresadas en incubadora por 24 días por bajo peso, Gemela 1: Peso:1.600 grs, presenta un mamelón preauricular y en las extremidades inferiores se visualizan manchas café con leche. Sostén cefálico a los 7 meses y Gemela 2: Peso: 1.570 grs. Sostén cefálico a los 6 meses. En ella la madre refiere crisis de hiperextensibilidad, sin diagnostico etiológico. Por lo demás el fenotipo de ambas es superponible: dolicocefalia, hipertelorismo, fisuras palpebrales estrechas con pliegues epicánticos. TAC, ecocardiograma y examen oftálmico normales para la edad. Hipotonía generalizada. Antecedentes familiares: madre y padre, casados en segundas nupcias, ambos de 44 años de edad, no consanguíneos. La madre tiene ocho hijos de su primera pareja y el padre un hijo. En el estudio citogenético de ambas se observó la presencia en mosaico de un cromosoma 20 en anillo: 45,XX,-20/46,XX,r(20) en el 5 % y 95% respectivamente de las metafases analizadas. Se desconoce reportes previos de ocurrencia del cromosoma 20 en anillo en gemelos monocigotos.
Discusión: Se discuten las posibles implicancias clínicas, considerando que esta anormalidad podría comprometer a dos genes relacionados con canalopatías epilépticas (CHRNA4 y KCNQ2).
Palabras Claves: cromosomas humanos Par20, cromosomas en anillo
ABSTRACT
Introduction: Ring chromosomes are an uncommon chromosomal disorder characterized by loss of the ends of the chromosome followed by fusing of the two ends to form a circle. The phenotype and clinical characteristics of the patient show a direct relationship with which chromosome is involved and the amount of genetic material lost from the ends. This abnormality can even be present without having a clinical effect. However, ring chromosome 20 syndrome is characterized by mental retardation, behavioral disorders, dysmorphism, and refractory epilepsy involving various types of seizures. It is caused by changes at the 20q13 region of chromosome 20 and is the third type of epilepsy known to be of genetic origin.
Case Report: Two female infants of 9 months of age presenting due to psychomotor retardation and dysmorphic features. Product of a monochorionic and monoamniotic twin pregnancy delivered by cesarean, both incubated for 24 days due to low birth weight; Twin 1, weighing 1,600 gm, presented a preauricular tag and café-au-lait spots on the legs; head raising at 7 months. Twin 2, weighing 1.570 gm; head raising at 6 months. The mother reports Twin 2 experienced hyperextension convulsions without causal diagnosis. Both otherwise present identical phenotypes with dolichocephaly, hypertelorism, narrow palpebral fissures with epicanthic folds. CT, echocardiogram, and ophthalmic exams normal for age. General hypotonia. Family history: Mother and father in second marriage, both 44 years of age, not consanguine. The mother has eight children by her first husband, and the father one child. Cytogenetic study of both showed a mosaic ring chromosome 20: 45,XX,-20/46,XX, r(20) in the 5% and 95% respectively of the metaphases analyzed. No previous reports of ring chromosome 20 in monozygotic twins appear in the literature.
Discussion: The possible clinical implications are under discussion since the abnormality could compromise the two genes related to epileptic channelopathies (CHRNA4 and KCNQ2).
Key words: chromosomes, human, Pair 20, ring chromosomes
INTRODUCCIÓN
El cromosoma en anillo es un aberración cromosómica poco frecuente, en su mayoría debido a la ruptura de ambos telomeros y posterior fusión de las porciones terminales delecionadas(1). El cromosoma 20 en anillo fue catalogado inicialmente en 1976 por Borgaonkar y col, como un síndrome genético, lo cual fue apoyado por Porfirio y Herva; y confirmado por Atkins, quién hizo la primera descripción del caso en un niño con retardo mental, problemas de desarrollo, crisis epilépticas y microcefalia asociada al cromosoma 20 en anillo (2-5).
Los pacientes portadores de una anomalía cromosómica como el cromosoma 20 en anillo tienen anormalidades neurológicas expresadas por un retardo mental moderado, retardo del desarrollo, diferentes tipos de crisis refractaria a la farmacoterapia, consistente en un estado prolongado de confusión con o sin crisis motora y dismorfias viscerales, usualmente renales y cardiacas. En muchos casos el desarrollo sicomotor suele normal durante la infancia hasta los dos años de edad y sin predominancia de sexo (3, 6-10).
A pesar de que los portadores presentan mosaicismos con presencia del cromosoma 20 en anillo que puede ir del 1 al 100%, un individuo puede ser considerado afectado por el síndrome del cromosoma 20 en anillo, cuando éste es observado en un porcentaje mayor al 50%, debido a que en un bajo porcentaje el fenotipo es menor. Estudios previos en series de pacientes concluyen que el radio de mosaicismo es inversamente correlativo al coeficiente intelectual (CI) y la edad de aparición (10-12).
En esta presentación, se describe un gemelar monocigoto con anillo del cromosoma 20.
REPORTE DEL CASO
Gemelar monocigoto monocorial monoamniotico del sexo femenino, de 9 meses de edad, referidas al Departamento de Genética del IICS para su estudio cromosómico, debido a un retraso en el desarrollo psicomotor, junto con dismorfias que se asemejaban al Síndrome de Down. Hijas de una pareja sana, no consanguínea, de madre y padre de 44 años, casados en segundas nupcias. La madre tiene ocho hijos de su primera pareja y el padre un hijo.
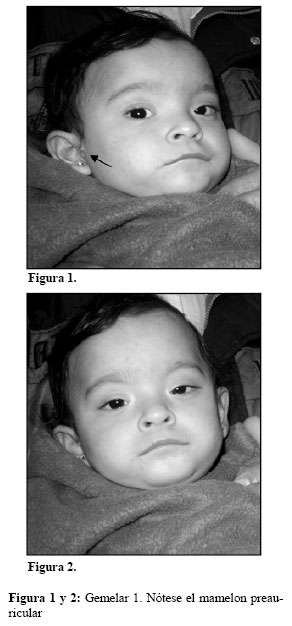
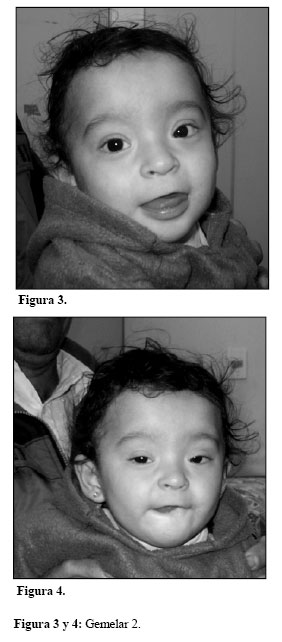
Sin historia familiar de epilepsia o anormalidades mayores en sus respectivas familias. Nacidas de un parto por cesárea, ambas ingresadas en incubadora por 24 días por bajo peso. Gemela 1: Peso al nacer 1.600 grs, presenta un mamelón preauricular y en las extremidades inferiores se visualizan manchas café con leche. Sostén cefálico a los 7 meses y Gemela 2: Peso al nacer 1.570 grs. Sostén cefálico a los 6 meses. En ella la madre refiere crisis de hiperextensibilidad sin diagnostico etiológico. Por lo demás el fenotipo de ambas es superponible: dolicocefalia, facies plana. Hipertelorismo ocular, fisuras palpebrales estrechas con pliegues epicánticos, inclinación mongoloide. Orejas despegadas, con implantación baja. Hipotonía generalizada. Tomografía Axial Computarizada (TAC), ecocardiograma y examen de ojos normal para la edad (Figura 1 y 2 Gemelar 1. Figura 3 y 4 Gemelar 2). En ambas obsérvese el hipertelorismo ocular, las fisuras palpebrales estrechas con pliegues epicánticos, inclinación mongoloide. Orejas despegadas con implantación baja.
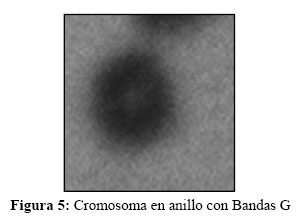
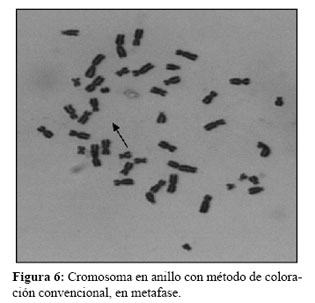
El análisis cromosómico fue llevado a cabo luego de un cultivo de linfocitos de sangre periférica en las gemelas y sus progenitores. Por cada paciente se realizaron tres cultivos diferentes en el tiempo de duración del cultivo así como en la extracción. Dos cultivos normales de 72 hs, uno se detuvo para observar células en metafase y otro en profase, para obtener cromosomas menos condensados y visualizar la formación en anillo (13) (Figura 5, 6 y 7).
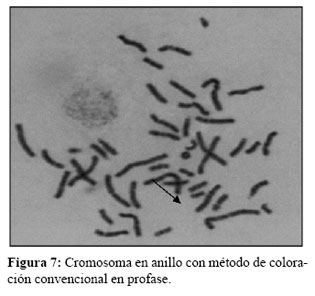
Un cultivo de 48 hs, debido a los cromosomas en anillo por su estructura muchas veces inestable tienden a no prosperar en cultivos de mayor tiempo.
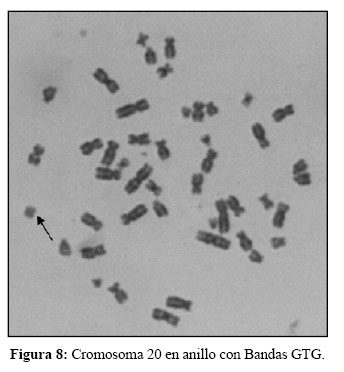
El complemento cromosómico en ambas gemelas fue de 45,XX,-20/46,XX,r(20)(p12;q13,2), en el 5% y 95% respectivamente de las metafases analizadas por niña, con los puntos de ruptura definidos por GTG (Figura 8).
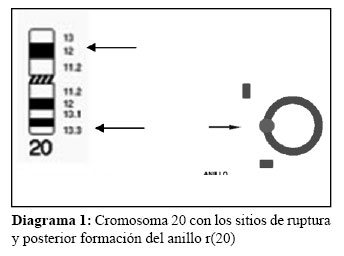
El r(20) tenía puntos de ruptura en el p13 y q13,33, formando un anillo (Diagrama 1).
No fueron observadas diferencias en los porcentajes de mosaicismos, entre individuos y entre los cultivos de 48 y 72 hs.
Ambos padres tenían un cariotipo normal. El cariotipo materno era 46,XX y el paterno 46,XY.
DISCUSIÓN
Se describe el caso de las gemelas monocigotas con un cromosoma 20 en anillo, de novo, considerando que no existen antecedentes familiares y ambos progenitores presentan cariotipo normal. Debido a que corresponden a gemelas monocigotas, monocorial, monoamnioticas, la formación del cromosoma en anillo debería haberse dado en una fase precoz antes de la separación del huevo fecundado.
El cromosoma 20 en anillo es uno de los síndromes asociados con episodios de epilepsia, además de problemas de desarrollo, problemas de aprendizaje, episodios de epilepsia resistentes al tratamiento con drogas antiepilépticas y de ausencia, así como un dimorfismo facial (9). Siendo el porcentaje de r(20) inversamente correlativo con la edad de aparición y el grado de CI. Las malformaciones mayores incluyendo anomalías cardiovasculares y renales con malformaciones menores malformaciones incluyendo dimorfismo facial y microcefalia fueron mas frecuentes en pacientes con un alto grado de mosaicismo(10,11).
Los síntomas clínicos de r(20) son causados por una parcial monosomía producida por la deleción de las regiones teloméricas. Recientemente ha sido reportado que la formación de un cromosoma en anillo es posible con un solo telomero roto (14). Pero se ha visto que la deleción de la porción telúrica del brazo corto del cromosoma 20 no cursa con epilepsia, si la deleción terminal del brazo largo que envuelve la región 20q13, da como resultado cuadros de epilepsia (15). A su vez la severidad de la expresión clínica ha sido descripta en relación directa con el tamaño del segmento delecionado en la región telomerica, lo cual fue enunciado por Serrano-Castro, enfatizando que el síntoma clínico mas común, la epilepsia, se halla determinada por la deleción que envuelve la región 20q13, como se menciona en varias revisiones (16). A pesar de todo el mecanismo del desarrollo de la epilepsia en el síndrome del r(20) aun permanece desconocido. Para lo cual existen descriptas dos posibilidades:1) una reducción de la proliferación celular durante el desarrollo cerebral, debido a la presencia de una anormalidad estructural y 2) la deleción de ciertos genes telomericos. La primera posibilidad es atendida teniendo en cuenta que en presencia de un cromosoma r(20) adicional no haya epilepsia y la segunda posibilidad ha sido soportada por el hecho de que los puntos de ruptura similares en la mayoría de los pacientes, incluyen una región telomerica 20q13,33, la cual incluye dos genes relacionados con canalopatías epilépticas, la epilepsia del lóbulo frontal nocturna autosómica dominante (canal para receptor neural nicotínico de la actilcolinesterasa) y las convulsiones neonatales benignas familiares (canales del K). Pudiendo ser estos dos locus fusionados en el cromosoma 20 en anillo, los que desaten la epilepsia o corresponder el r(20) a un tercer tipo de epilepsia.
La posibilidad de predicción de la clínica, así como el control de las niñas detectadas, es muy importante a la hora del seguimiento de las pacientes, pues el síndrome del cromosoma 20 en anillo muestra un deterioro gradual a nivel cerebral en caso de no tener un diagnostico y tratamiento. De ahí que se propone que el estudio cromosómico podría ser uno de los exámenes de primera línea en pacientes con epilepsia o ausencias con o sin problemas de aprendizaje o con mínima dismorfología. Ya que el establecimiento de una razón etiológica definitiva es muy importante para los profesionales de la salud como para los padres, para el tratamiento como para el asesoramiento genético.
REFERENCIAS
1. Kosztolanyi G. Does ring syndrome exist?: an análisis of 207 cases reports ons patients with a ring autosome. Hum Genet. 1987;75:174-179. [ Links ]
2. Borgaonkar DS, Lacasste YR, Stoli O. Usefulness of chromosome catalog in delineating new syndromes. Birth Defects. 1976;12:87-95. [ Links ]
3. Porfirio B, Valoran MG, Giannotti A, Sabetta G, Dallapiccola B. Ring 20 chromosome phenotype. J Med Genet. 1987;24:375-377. [ Links ]
4. Herva R, Saarinen J, Leikkonen L. Ther(20) syndrome. J Med Genet. 1977;14:281-283. [ Links ]
5. Atkins L, Miller WL, Salam M. A ring 20 chromosome. J Med Genet. 1972;9:377-380. [ Links ]
6. Burnell RH, Ster LM, Sutherland GR. A case of ring 20 chromosome with cardiac and renal anomalies. Aust Paediatr J. 1985;21:285-286. [ Links ]
7. Holopainen I, Penttinen M, Lakkala T, Aarima T. Ring chromosome 20 moisacism in a girl with complex partial seizures. Dev Med Child Neurol. 1994;36:70-83. [ Links ]
8. Da Mota-Gomes M, Lucca I, Monteiro-Bezerra AS, LLerena J, Madeira-Moreira D. Epilepsy and ring chromosome 20: case report. Arq Neuropsiquiatr. 2002;60(3A):631-35. [ Links ]
9. Alpman A, Serdaroglu G. Ring chromosome 20 syndrome with intractable epilepsy. Developmental Medicine & Child Neurology. 2005;47:343-346. [ Links ]
10. Nishiwaki T, Hirano M, Kumazawa M, Ueno S. Mosaicism and phenotype in ring chromosome 20 syndrome. Acta Neurol Scand. 2005;111:205-208. [ Links ]
11. Augustun PB, Parra J, Wouthers CH, Joosten P, Lindhout D, van EmdeBoas W. Ring chromosome 20 epilepsy syndrome in children: electronical features. Neurology. 2001;57:1108-1111. [ Links ]
12. Yamadera H, Kobayashi K, Suga H, Kaneko S. A study of ring 20 chromosome karyotype with epilepsy. Psychiatry Clin Neurosci. 1998;58:63-68. [ Links ]
13. Verma RS, Babu A. Human chromosomes. Manual of Basic Tecnhiques. New York: Pergamon Press; 1989. [ Links ]
14. Garcia-Cruz D, Vasquez AI, Perez-Rulfo D, Davalos NO, Peñaloza JM, Garcia-Ortiz JE, et al. Ring 20-syndrome and loss of telomeric regions. Ann Genet. 2000;43:113-116. [ Links ]
15. Phillips HA, Sceffer JE, Berckovic SF, Hollway GE, Sutherland GR, Mulley JC. Localization of a gene for autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q13,2. Nat Genet. 1995;10:117-118. [ Links ]
16. Serrano-Castro PJ, Aguilar-Castillo MJ, Olivares-Romero J, Jimenez-Machado R, Molina-Aparicio MJ. Ring chromosome 20: an epileptic channel disorder?. Rev Neurol (Barc). 2001;32:237-241. [ Links ]

