










Serviços Personalizados
Journal

Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPediatría (Asunción)
versão On-line ISSN 1683-9803
Pediatr. (Asunción) v.33 n.2 Asunción dez. 2006
CASO CLINICO
Duplicación distal del brazo largo del cromosoma 3 en línea pura: a propósito de un caso
Distal duplication of the long arm of chromosome 3
Elodia Torres, Marta Ascurra, Norma Monjagata, Stella Rodríguez.*
* Departamento de Genética. Instituto de Investigaciones en Ciencias de la Salud. Universidad Nacional de Asunción
Solicitud de Sobretiros: Elodia Torres. Rio de la Plata y Lagerenza. IICS. Barrio Sajonia.Asunción.
RESUMEN
Se reporta el caso de una niña de 25 días de vida con una duplicación distal del brazo largo del cromosoma 3, lo cual ocasiona una trisomía 3q2, enfermedad cromosómica muy rara. Los síntomas más importantes que presentan estos individuos, incluyen entre otros, retraso pre y post natal del crecimiento, retraso mental severo, microcefalia, sinofrismo, hipertricosis, malformaciones del corazón, de los riñones y de otros órganos internos y ocasionalmente anomalías de los miembros. El estudio cromosómico se llevó a cabo en sangre periférica, con técnicas de coloración convencional y de identificación con Bandas G y C. El cariotipo de la paciente resultó: 46,XX,dup3(q25 qter). El análisis cromosómico de los padres fue normal. Las características clínicas observadas inicialmente en la niña fueron compatibles con el Síndrome de Beckwith-Wiedeman y el de Cornelia de Lange, descartados a través del estudio citogenético, es por ello que se resalta la importancia del estudio cromosómico en el diagnóstico diferencial de síndromes de origen génico, para el diagnóstico de certeza y el asesoramiento genético a los padres.
Palabras claves: Duplicación 3q, síndrome dup (3q), cromosoma 3.
SUMMARY
This report is on a girl of 25 years old with a distal duplication of the long arm of chromosome 3. Trisomy 3q is a rare disease caused by the duplication of the long arm of chromosome 3. The most important signs of the disease includes pre and post-natal growth delay, severe mental retard, microcephaly, synophrism, hypertrichosis, heart, kidney and other organs malformations among others and occasionally limbs anomaly. The chromosome study was carried out in peripheral blood with conventional staining techniques andGC and C band identification. The cariotype of the patient was 46, XX, dup 3 (q25 qter) while the cariotypes of the parents were normal. The clinical characteristics were also compatible with Beckwith-Wiedeman and Cornelia de Lange syndromes that were discarded by chromosome analysis. We highlight in this paper the importance of making an accurate chromosome study in the differential diagnosis of genetic origin syndromes for the certainty diagnosis and genetic counseling to parents.
Key words: 3q duplication, dup(3q) syndrome, chromosome 3.
INTRODUCCION
Si bien, la duplicación en línea pura de todo el brazo largo del cromosoma 3, es muy rara y muy pocos casos han sido reportados (1), la duplicación de la porción terminal 3q2, ha sido descrita por Falek desde 1966, siendo inicialmente confundidos con el Síndrome de Cornelia de Lange, debido a la similitud en las manifestaciones clínicas que presentaban. Recién en el año 1973 con el advenimiento de las técnicas de bandeo cromosómico, se puso en evidencia la presencia de la duplicación de la porción terminal (3q2). Constituyéndose en una entidad separada y asociada a un fenotipo característico, entre los que se incluyen rasgos faciales típicos como microcefalia, sinofrismo, orejas de implantación baja, hipertricosis, malformaciones de manos y pies; defectos genitourinarios, anomalías cardíacas congénitas en combinación con retardo mental y del crecimiento. Alrededor del 1/3 de los pacientes no sobreviven al primer año de vida, principalmente por las malformaciones cardíacas y la susceptibilidad a infecciones (1-3).
La mayoría de los casos descritos con el síndrome de la duplicación 3q2, fueron el resultado de translocaciones desbalanceadas (1), originadas por recombinación desigual o por segregación anormal meiótica en un portador de una translocación balanceada o una inversión del cromosoma 3 (4). Unos pocos casos de aparición de novo fueron reportados en presencia de una inversión pericéntrica del cromosoma 9 ó por una inserción invertida del brazo corto del mismo cromosoma (5-8).
Se reporta el caso de una niña que es remitida a la consulta por un fenotipo sindromático, compatible con el Síndrome de Becwith-Wiedeman y de Cornelia de Lange.
DESCRIPCION DEL CASO CLINICO:
M.A.V.F. de sexo femenino, producto de tercer embarazo, padre de 24 años y madre de 22 años. La madre refiere prenatal controlado; parto cesárea por macrosomía. Peso al nacimiento 4000gs, talla 50cm, cc 37cm. Padres no consanguíneos y sin antecedentes familiares.
A las 24 horas de vida es derivada al servicio de pediatría por la facies compatible con el Síndrome de Beckwith-Wiedeman o el de Cornelia de Lange, con onfalocele, probable traumatismo obstétrico, hipoglicemia persistente, hipertricosis, sin convulsiones, ni ictericia. La ecografía transfontanelar y la tomografía de cráneo reportaron agenesia de cuerpo calloso; por ecocardiografía se detectó CIV muscular ubicado en tercio inferior del septo interventricular (CIV 3mm), y por ecografía abdominal se informó asas dilatadas sin líquido en cavidad. A los 14 días de vida se le realizó el cierre primario del onfalocele, permaneció internada durante 25 días y es dada de alta y remitida al Departamento de Genética del IICS, con el siguiente diagnóstico: RNT/GEG, facies sindromática, onfalocele, infección urinaria y agenesia de cuerpo calloso. La niña fallece por neumonía a los diez meses de edad.
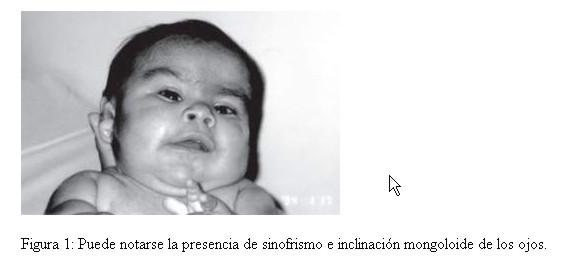
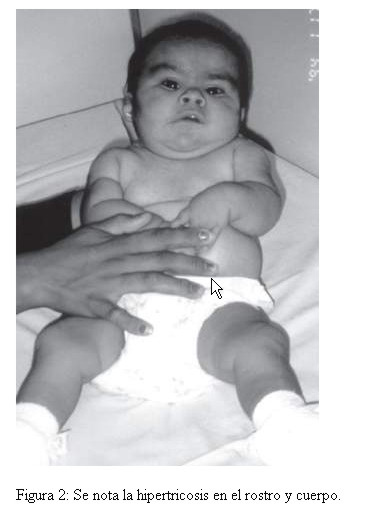
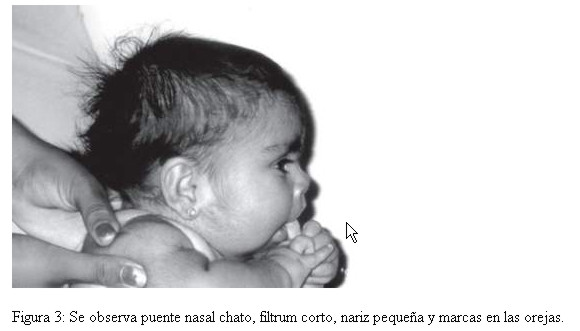
Por las características clínicas, entre las que se destacan, fontanelas amplias, cejas pobladas, sinofrismo, puente nasal chato e hirsutismo, se sospecho como portadora de un Síndrome de Cornelia de Lange, y por la presencia de hemangioma plano en frente orificio preauricular, narinas antevertidas, labio en arco de cupido, micrognatia, hipoglicemia, onfacele y macrosomia, de un Síndrome Becwith-Wiedeman, además de cuello corto, nariz pequeña, marcas en las orejas y filtrum corto, fosita coccigea profunda; pliegue palmar único en ambas manos y clinodactilia; pliegues plantares en el medio (Fig. 1, 2 y 3). Con estos antecedentes, se solicitó el estudio citogenético para descartar alteraciones cromosómicas, asociadas al fenotipo descrito.
El estudio cromosómico tanto de la niña como de sus progenitores, se realizo a través del cultivo de linfocitos de sangre periférica, en medio RPMI enriquecido con 15% de suero fetal bovino y fitohemaglutinina (Mform, liofilizado), a 37º durante 72 horas. Transcurrido el tiempo se agregó colchicina 0,003 ug/ml durante 90 minutos, posterior tratamiento con solución hipotónica KCl 0,075M, y fijación con Carnoy I (3:1) metanol: ácido acético. Se realizaron los extendidos sobre portaobjetos, los cuales se tiñeron con Giemsa para el estudio convencional, la identificación cromosómica se llevó cabo con las técnicas de bandas G y C (8,9).
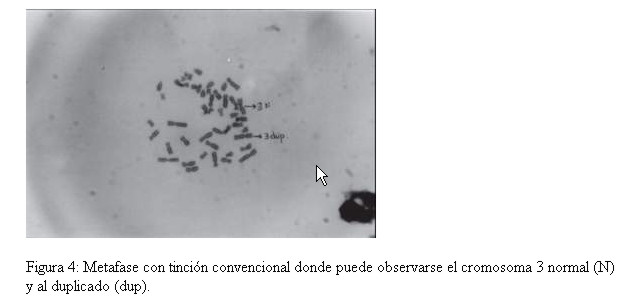
Fueron estudiadas cincuenta células por paciente. Encontrándose en todos ellos, con tinción convencional 46 cromosomas y con la técnica de Bandeo G, se detectó en todas las metafases analizadas de la niña, la presencia de una duplicación del brazo largo de uno de los cromosomas del par 3 región q25 hasta la porción terminal (qter) (Fig. 4 y 5) Ninguna anomalía fue observada en los cromosomas de los progenitores. Cariotipo de la niña: 46,XX, dup(3)(q25 qter) Cariotipo de la madre: 46,XX Cariotipo del padre: 46,XY
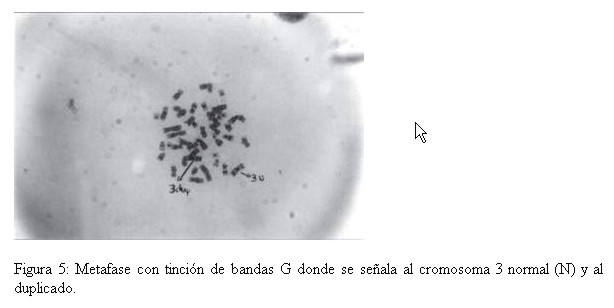
DISCUSION
La duplicación del cromosoma 3q2 es considerado raro, la mayoría de los pacientes reportados con este síndrome, son productos de padres portadores de trans-locaciones balanceadas (1). Se reporta un caso de novo, de la duplicación 3q25 a la porción 3qter en línea pura, probablemente debido a una falla en la recombinación meiótica en uno de los progenitores.
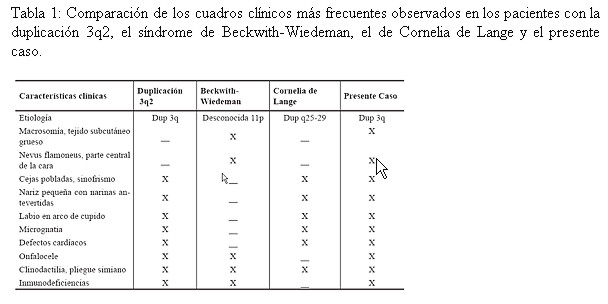
La niña que presentó inicialmente un fenotipo compatible con el síndrome de Beckwith-Wiedeman y con el de Cornelia de Lange (Tabla 1), fueron descartados por un diagnóstico diferencial a través del análisis citogenético, con lo cual se confirmó el Síndrome de la Duplicación 3q2 en la niña. De hecho, el locus 3q26 para el brazo largo del cromosoma 3, según el cuarto Taller Internacional en el mapeo de este cromosoma realizado en el año 1993 (10), se denominó CDL y corresponde precisamente al gene del Síndrome de Cornelia de Lange.
Queda confirmada de esta manera la importancia de la realización del estudio cromosómico a pacientes portadores de señales clínicas compatibles con síndromes de origen génico, para el diagnóstico de certeza y el asesoramiento genético a los padres.
REFERENCIAS BIBLIOGRAFICAS
1. Faas BH, De Vries BB, Van Es-van Gaal J, Merkx G, Draaisma JM, Smeets DF. A new case of dup(3q) syndrome due to a pure duplication of 3qter. Clin Genet. 2002;62(4): 315-20. [ Links ]
2. Jones KL. Smith´s Recognizble Patterns of Human Malformation. 5th Edition. Philadelphia – USA: Saunders Company; 1997. [ Links ]
3. Gorlin RJ, Cohen M, Michael JR, Hennekam RCM. Syndromes of the Head and the Neck. 4th Edition. USA: Oxford University Press; 2001. [ Links ]
4. Thompson T. Genética en Medicina. 4ª Edición. Barcelona: Masson SA; 1996. [ Links ]
5. Ayral D, Raudrant D, Charleuz JP, Noel B. Duplication of the long arm of chromosome 3 (dup3q) in a newborn infant whose the father is carrier of pericentric inversion of chromosome 9. Pediatric. 1984;39(8):681-90. [ Links ]
6. Zenteno JC, Pérez A, Rivera MR. Identificación de una nueva anomalía citogenética en el síndrome de duplicación 3q. Rev Med Hosp Gen Mex. 1997;60(4):218-20. [ Links ]
7. Preus M, Vekemans M, Kaplan P. Diagnosis of Chromosome 3 Duplication q23àqter, Deletion p25àpter in a Patient With the C (Trigonocephaly) Syndrome. Am J Med Genet. 1986;23(4):935-43. [ Links ]
8. Wilson GN, Dasouki M, Barr M Jr. Further Delineation of the dup(3q) Syndrome. Am J Med Genet. 1985;22(11):117-23. [ Links ]
9. Verma RS, Babu A. Human Chromosomes: Manual of basic techniques. New York: Pergamon Press;1989. [ Links ]
10. Naylor SL, Buys CH, Carritt B. Report and abstracts of the Fourth Internacional Workshop on Human Chromosome 3 Mapping. Cytogenet Cell Genet.1994;65(1-2):2-50. [ Links ]

