











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La histiocitosis de células Langerghans (HCL) es un tipo de histiocitosis caracterizada por la presencia de células del mismo nombre. Estas son células dendríticas del sistema mononuclear fagocítico derivadas de la médula ósea, pero llevan el nombre de Langerhans por la similitud de las células de Langerhans de la piel, los progenitores de estas últimas son células del saco vitelino y de los monocitos derivados del hígado fetal que pueblan la piel antes del nacimiento y se mantienen localmente en condiciones de estado estable, en cambio las células derivadas de monocitos en sangre periférica migran a la epidermis tras el proceso de inflamación y se diferencian en células similares a las células de Langerhans1.
La HCL comparte marcadores de superficie con las células de Langerhans epidérmicas (CD1a+ / CD207+) y una expresión aumentada de genes asociados con precursores de células dendríticas mieloides inmaduras. Badalian-Very y sus colegas identificaron la mutación BRAF V600E en más de la mitad de las lesiones de HCL, 40 un hallazgo que se verificó posteriormente en otras series y se atribuyó a la célula de HCL patógena2,3.
La proteína BRAF es un miembro de la familia RAF de serina / treonina quinasa, y es un componente clave de la vía de señalización MAPK (RAS-RAF-MEK-ERK) que conduce a la activación de factores de transcripción esenciales para el crecimiento y la proliferación celular. La transversión V600E (c.1799T> A en el exón 15) es la mutación más común en BRAF y es uno de los principales impulsores de las neoplasias malignas humanas que conducen a la activación constitutiva aguas abajo de MEK y ERK. Los estudios han informado que BRAF-V600E se observa con más frecuencia en pacientes con HCL multisistémica que en pacientes con enfermedad aislada y esto Se asocia con un mayor riesgo de recaída4.
Se han confirmado otras mutaciones en los genes de la vía MAPK en casi el 80% de los pacientes con HCL. Se podrían descubrir mutaciones adicionales de MAPK porque la vía ERK se activa en el 100% de los pacientes con HCL y es así que la HCL tambien puede presentarse junto a otros canceres histológicos con mutaciones compartidas, como el caso de cancer de tiroides5.
La incidencia anual de HCL es de 4,6 casos por 1 millón de niños menores de 15 años, con una proporción de hombres a mujeres de 1,2: 1 y la incidencia estimada entre adultos es de 1 a 2 casos por millón6,7. Registros en EEUU han observado mayor incidencia en raza hispana que afroamericanos8.
La presentación clínica de esta patología es muy variable y puede ir desde lesiones asintomáticas únicas hasta una enfermedad multisistémica. El hueso y la piel tienen una mayor frecuencia de afectación, así como el eje hipotalámico hipofisario, aunque también se pueden ver alteradas otras glándulas endocrinas, pero en menor medida, por ejemplo, la glándula tiroides. Los niños con afectación del hígado, el bazo o la médula ósea tienen un desenlace fatal, por lo tanto, se clasifican como pacientes con HCL de alto riesgo6. Se han descrito casos en que la presentación inicial fue en un solo órgano, como en el caso de la hipófisis o de la tiroides causando una diabetes insípida o un hipotiroidismo respectivamente y con el tiempo evolucionan a afectación sistémica9. En los adultos, la HCL pulmonar aislada (PLCH) es la manifestación más común y se considera un tipo específico de HCL por su origen policlonal10.
Histológicamente se observan lesiones características granulomatosas destructivas que contienen células mononucleares con núcleos dentados, estas son las células de Langerhans presentadoras de antígenos dendríticos (con gránulos de Birbeck), también existe infiltración de linfocitos y granulocitos eosinofílicos y la Inmunohistoquimica presenta positividad para S100, CD1a y Langerina (CD207)11. El aspecto histológico benigno de la célula CD207+, el infiltrado inflamatorio que la acompaña y la tormenta de citocinas tanto local como sistémica respaldan un origen inflamatorio de la HCL, mientras que la clonalidad, las mutaciones del gen activador somático en la vía de la proteína quinasa activada por mitógenos (MAPK) y las mutaciones compartidas con precursores hematopoyéticos favorecen la reclasificación de la HCL como un trastorno neoplásico mieloide4. Durante la Revision de Clasificaciones de la Sociedad de Histiocitos en el 2016 se redefinió a la HCL como una neoplasia mieloide inflamatoria11. En la práctica profesional el diagnostico no está exento de dificultades debido a la naturaleza poco frecuente y las diferentes formas en cuanto a presentación clínica, particularmente entre pacientes adultos; los estudios auxiliares, imagenológicos y la histología tienen un papel fundamental tanto para el diagnóstico como para el seguimiento sobre todo en pacientes con riesgo de una evolución tórpida o complicación sistémica. Finalmente traemos a colación el caso de un adulto, con HCL localizado en la glándula tiroides, cuyo diagnóstico se realiza luego de una tiroidectomía total.
CASO CLINICO
Varón, 61 años, sin antecedentes patológicos de valor, consulta por dificultad para deglutir desde hace algunos meses. Examen físico: tiroides aumentada, palpación de nódulos en ambos lóbulos, consistencia firme, no adheridos a estructuras vecinas. Laboratorio: Perfil tiroideo normal. Rutina normal. Ecografía: bocio multinodular a expensas de nódulos hiperecogénicos de 31.5x19 mm y 49.7x29.6 mm y adenomegalia cervical bilateral. Se realiza una punción aspiración con aguja fina (PAAF) del nódulo tiroideo izquierdo; que informa lo siguiente: “Extendido hipercelular con numerosas placas, acúmulos tridimensionales y células aisladas con atipia nuclear, así como numerosas células multinucleadas en un fondo hemorrágico, categoría Bethesda III; atipia de significado indeterminado”, como pie de nota se sugiere que podría tratarse de un Carcinoma Papilar de Tiroides, pero el material presenta severos artefactos de fijación y aplastamiento que dificultan la evaluación. Por tanto, se decide realizar una tiroidectomía total y biopsia de ganglios. La anatomía patológica informa: “Parénquima extensamente reemplazado por una proliferación celular; aspecto histiocítico, células grandes y medianas, citoplasma amplio claro, y eosinofílico con hendiduras nucleares; abundante población eosinofílica. El aspecto histológico es inusual y el diagnóstico diferencial se establece entre una Histiocitosis de Células de Langherans, un Linfoma de Hodgkin y una Tiroiditis Granulomatosa. Ganglios peritiroideos con hitiocitos en senos cortical y medular” (Figuras 1 y 2).
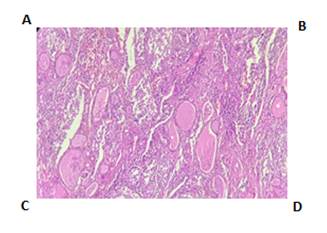
Figura 1. Anatomía Patológica. Coloración H y E. A: Imagen histológica a 40x, se observa el tejido tiroideo (folículos) parcialmente reemplazado por la lesión con vaga nodularidad. (Mitad izquierda tiroides poco conservada-mitad derecha lesión). B: Imagen histológica a 100x, folículos tiroideos y lesión. C: Imagen histológica a 400x, donde se observa infiltrado mixto de células mononucleares con citoplasma eosinófilo, núcleo grande, algunas de ellas con hendiduras. Se nota también gran cantidad de eosinófilos. D: Imagen histológica a 400x. Se nota también una célula gigante multinucleada.
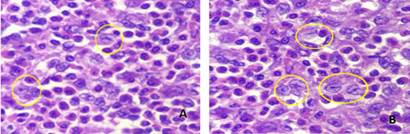
Figura 2. Células histiocíticas/eosinofilicas. Coloración H y E. A y B: Hay numerosas células de Langerhans (núcleo redondo u ovalado, membrana nuclear nítida, cromatina fina con hendidura o nucleolo pequeño), solo algunas están marcadas con el círculo amarillo.
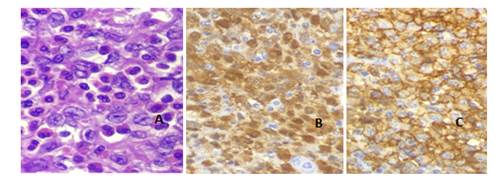
Tras la Inmunohistoquímica (Fig 3) se observan células LCA+, S100+, CD1a+ CD68+/- (Fig 3). DIAGNOSTICO FINAL: Histiocitosis de Células de Langerhans. TAC sin otras lesiones focales, no adenomegalias. Tratamiento levotiroxina. Seguimiento por hematología, no requirió quimioterapia, actualmente libre de enfermedad.
DISCUSION
La afectación de la hipófisis posterior es la presentación endocrina más frecuente de HCL, particularmente en niños6. Las lesiones en la glándula tiroides son extremadamente raras y el compromiso único de tiroides lo es aún más; con prevalencia en mujeres adultas, presentándose como agrandamiento difuso más que nodular y sin síntomas, a diferencia de este caso. La revisión hecha por Patten y col en el 2012, notifica 75 casos de afectación de la glándula tiroides por HCL, la mayoría de la literatura inglesa; 59% de los cuales presentaba bocio, el 25,8% nódulos tiroideos, el 40,9% de los pacientes estaban en eutiroidismo, seguido por hipotiroidismo e hipertiroidismo subclínico. En la mayoría de los casos la afectación de la tiroides fue parte de una enfermedad multisistémica, predominando en adultos más que en niños, con una proporción de mujeres a hombres ligeramente superior (1,4: 1 respectivamente). El 15% de los que tenían afectación tiroidea estaba asociadoa HCL en hipófisis posterior12.
La HCL puede co-existir junto con otras patologías de la glándula tiroides. El primer caso notificado de Bocio, Carcinoma Papilar de Tiroides (CPT) y HCL simultáneas fue en un paciente con HCL de larga duración. Se ha sugerido que ambos procesos podrían tener vías comunes de patogénesis, y que el proceso inflamatorio subyacente y la mutación de BRAF también podrían desencadenar el desarrollo de CPT5. Recientemente Kim Ik y colaboradores tras el diagnóstico de un caso de HCL en cráneo, asociado a un carcinoma papilar de tiroides y Enfermedad de Castleman, realizan una revisión en la literatura, encontrando 19 casos de HCL en tiroides y carcinoma papilar, 2 de ellos en niños, la edad media de los adultos fue de 36 años, 11 eran hombres, 4 casos se presentaron en forma metacrónica y 5 con lesiones en ganglios, sólo 8 caso tenía extensión a otros órganos13. Se han descrito también otras patologías concomitantes de la tiroides como la enfermedad de Graves e hipotiroidismo central o primario, pero en general son eutiroides como este paciente14.
Para la presentación de este caso, se buscó publicaciones en Pub Med de casos relacionados a HCL y tiroides, del 2013 a 2020, en inglés y español y se encontraron 25 casos de los cuales 3 eran niños, la edad promedio en adultos fue 40 años, la relación hombre/mujer fue de 1.5/1, la presentación clínica más frecuente fue de un bocio nodular o multinodular (20 casos), a diferencia de lo que publicó Patten12. Nueve casos se presentaron como una tiroiditis dolorosa, 4 con hipotiroidismo y 2 con hipertiroidismo. Sólo se encontraron 2 casos más publicados con CPT. Trece tenían afectación de otros órganos y en 8 pacientes fueron diagnosticados de una Diabetes Insípida (Cuadro 1).
El presente caso se trata de un varón, cuyos síntomas compresivos lo llevan a la consulta, en donde se constata luego de una Ecografía un bocio multinodular, pero con una punción un poco confusa ya que finalmente informa como un Bethesda III, sin descartar la posibilidad de un carcinoma papilar. En muchos casos el diagnóstico diferencial de la HCL que afecta a la tiroides no resulta evidente porque los signos y síntomas son variables e inespecíficos. y se realiza luego del estudio de la pieza quirúrgica, como en esta ocasión15, En particular, los nódulos tiroideos o el bocio difuso pueden diagnosticarse erróneamente como las siguientes enfermedades: tumores benignos, carcinoma indiferenciado, linfoma, tiroiditis linfoide, tiroiditis granulomatosa crónica, la aspiración con aguja fina es esencial para la HCL16.
En el caso actual, la PAAF se describe característicamente como un extendido hipercelular, con numerosas células gigantes multinucleadas. Es necesaria una alta sospecha para guiar al diagnóstico final; en algunos pacientes aun luego de una tiroidectomía y una biopsia formal, se confunden los diagnósticos con una tiroiditis subaguda y al presentarse otros síntomas de afectación extratiroidea (diabetes insípida, lesiones líticas oseas), se realiza el diagnostico restrospectivamente tras revisión de placas e inmunohistoquimica9.
El paciente fue sometido a una tiroidectomía total confirmándose el diagnóstico de HCL con afectación de los ganglios peritiroideos, a través de la Anatomía Patológica y la Inmunohistoquímica en donde se observan las células características LCA+, S100+, CD1a+ CD68+/-. (fig 1, 2 y 3).
Cabe destacar que también están indicados los estudios genéticos ya que la presencia de mutaciones BRAF-V600E implica un riesgo de enfermedad multisistémica y de recaída4, pero no se pudo realizar en el paciente por razones de disponibilidad.
Si bien no existen ensayos prospectivos en cuanto a un tratamiento estándar, es fundamental conocer el grado de afectación de órganos para decidir el manejo; teniendo en cuenta la siguiente clasificación: HCL de sistema único (SS-HCL) con afectación multifocal o unifocal, o como HCL multisistémica (MS-HCL) con afectación de múltiples órganos con o sin afectación de órganos de riesgo1,17. La enfermedad extendida al sistema hematopoyético es extremadamente rara en adultos y si el bazo, hígado o Sistema Nervioso Central están implicados, esto lleva a un pronóstico desfavorable si no hay respuesta al tratamiento. La fiebre, los sudores nocturnos y la pérdida de peso podrían predecir el curso agresivo de HCL raramente observado en adultos comparable al del linfoma no Hodgkin de alto grado17. Para los casos de afectación sistémica se opta por quimioterapia y corticoides, incluyendo las terapias dirigidas como el Imatinib o Vemurafenib, estos últimos muestran efectividad en casos con mutación BRAF-V600E18-20.
Para los casos locales o de un sólo órgano la conducta es aún más discutida, pero se tienen datos de que la enfermedad aislada en el adulto tiene un buen pronóstico21,22. Se describen casos sobre todo en tiroides, con buena respuesta y sin evolución a otros tejidos luego de cirugía sola, principalmente en aquellos de hallazgo incidental23. El compromiso aislado de los ganglios linfáticos es raro y se han descrito regresiones espontáneas, por lo que el vaciamiento ganglionar extenso no está indicado24.
Cuadro 1. Casos publicados desde 2013 al 2020
| Autores (Año) | Edad/Sexo | Características clínicas y estado funcional de tiroides | Afectación de otros órganos |
|---|---|---|---|
| Ozisik et al (2020)26 | 58/M | Nódulo solitario (NS) | Hipófisis |
| 45/M | 2 nódulos tiroideos | Hipófisis (Panhip, DIC). Ganglios cervicales. Mucosa gingival. | |
| Ben Nacef et al. (2020)9 | 34/F | Bocio difuso doloroso | Hipófisis. (Panhip, DIC) |
| He et al. (2020)27 | 3,5/M | Masa difusa. Obstrucción traqueal. Hipotiroidismo | Ganglios cervicales, pulmones, hígado, bazo, piel. |
| Xie y col (2018)28 | 41/M | Bocio Multinodular (BMN). | Hipófisis (DIC). Hígado. |
| Kuhn y col (2016)29 | 73/M | NS. Hipotiroidismo subclínico | |
| Skuwrosnska (2016)30 | 39/F | BMN. Tiroiditis dolorosa | |
| Saqi (2015)15 | 35/F | NS | Hipófisis. (Panhip, DIC). |
| Chrisoulidou (2015)14 | 26/M | BMN. Hipertiroidismo | |
| Attakkil (2015)31 | 8/M | BMN | |
| Sangtian (2015)32 | 57/F | BMN | Hipófisis (Panhip, hiperprolactinemia DIC). |
| Roy (2015)33 | 13/M | NS. Compresión traqueal. Tiroiditis dolorosa. | |
| Long (2015)34 | 35/M | BMN. Tiroiditis dolorosa | Hipófisis (DIC). |
| Ozan (2015)35 | 6/f | BMN | |
| 38/M | BMN | Hipófisis (DIC). Medula ósea. | |
| 9/M | BMN | ||
| Cai (2015)36 | 24/M | BMN. Tiroiditis dolorosa | Cuerpos vertebrales s1-s2 |
| Diego E (2014)37 | 33/M | NS. Tiroiditis. Hipotiroidismo | Hipófisis (DIC, HH) pulmón, |
| Marupudi (2014)38 | 33/M | BMN doloroso. Hipotiroidismo. | Ulcera paladar |
| Chen (2014)39 | 27/M | Masa cervical, nódulos tiroideos. Tiroides dolorosa. Hipertiroidismo subclínico | |
| 38/F | Nódulos. tiroides dolorosa. Hipertiroidismo subclínico | ||
| Znati (2013)40 | 52/M | Bocio difuso | |
| Pusztaszeri (2013)41 | 25/F | NS. Tiroiditis dolorosa |
Panhip: panhipopituitarismo, DIC: Diabetes Insípida Central, HH: hipogonadismo hipogonadotrofico
En la revisión de Patten la mayoría de los niños se sometieron a una combinación de cirugía y quimioterapia para el tratamiento de la HCL tiroidea (52,6%), mientras que la mayoría de los adultos se sometieron a cirugía sola (57,5%), seguida de cirugía y quimioterapia o radioterapia (21,3%)12.
Tras el diagnóstico postoperatorio, el paciente fue evaluado por Hematología, quienes no encontraron datos clínicos ni imagenológicos de una enfermedad sistémica, por lo que se decide espectar el tratamiento quimioterápico, tampoco se encontraron deficiencias hormonales ni diabetes insípida en la evaluación endocrinológica. Por otro lado, el seguimiento periódico es fundamental para descartar recurrencia en otros órganos y sistemas, inclusive la aparición de cáncer en otros tejidos25.
En conclusión, la HCL es una neoplasia rara y el compromiso único de tiroides es aún más, con prevalencia en mujeres adultas, presentándose como agrandamiento difuso más que nodular y sin síntomas, a diferencia de este caso. Existen reportes relacionados con cáncer papilar de tiroides, enfermedad de Graves e hipotiroidismo central o primario, pero en general son eutiroides como este paciente. También se reporta como primera manifestación de enfermedad sistémica, por lo que es importante el seguimiento. La PAAF raramente es concluyente, el diagnóstico definitivo es histológico.

