











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introducción
A nivel mundial, la esclerodermia es una enfermedad cuya incidencia afecta 4-12 casos/millón de habitantes/año. Predomina en mujeres. Edad de comienzo es 40 años, sin preferencia étnica y distribución universal. La piel se vuelve brillante y dura. Existen dos tipos: esclerodermia localizada y esclerosis sistémica1,2.
La esclerodermia localizada (LS) es un espectro de enfermedades que afectan principalmente a piel. LS puede involucrar tejidos adyacentes (grasa, fascia, músculo y hueso, pero no órganos internos). Incidencia de 0,4-2,7/100 000 personas y 2,6-6 veces más en mujeres. Morfea, el subtipo más frecuente aparece en adultos entre 40 y 50 años. La LS presenta una clasificación que considera la extensión y profundidad de la fibrosis y comprende cinco tipos principales y ciertos subtipos3.
Histológicamente, la LS presenta engrosamiento y homogenización de fibras de colágeno, con infiltrado perivascular de tipo linfohistiocítico, infiltrado mayor en tejidos subcutáneos donde el tejido adiposo es reemplazado por colágeno. La dermis se presenta edematosa, en etapa tardía el tejido inflamatorio desaparece y las bandas de colágeno se vuelven más gruesas y densas. Los folículos pilosos, glándulas sudoríparas y tejido graso subcutáneo se pierden y el colágeno se acumula4-6.
Se debe considerar una variedad de diagnósticos diferenciales en LS. Lo primordial es diferenciarlo con la esclerodermia sistémica debido a su pronóstico. La afectación de la piel de zonas acrales, ausencia del fenómeno de Raynaud y/o síntomas sistémicos y la preservación de la microcirculación cutánea a nivel periungueal, indican que el cuadro es localizado. Sin embargo, las lesiones tempranas extensas o generalizadas plantean dudas; evolución del paciente y exámenes complementarios son de gran utilidad7-9.
Aunque no existe tratamiento causal para LS, existe variedad de terapias terapéuticas disponibles, especialmente para fase activa de la enfermedad. Las opciones de tratamiento para LS podrían dividirse en tópicos y terapia sistémica, así como fototerapia ultravioleta (UV). El alcance y gravedad de LS deben tenerse en cuenta antes de iniciar la terapia3.
En Paraguay no existe un registro de pacientes con Morfea y la incidencia de la misma, razón por la cual representa un tema de salud pública que debe ser estudiado y expuesto para el conocimiento de la población y velar por la salud de estos.
PRESENTACIÓN DEL CASO
Paciente de sexo femenino de 23 años de edad que consulta en el Hospital de Clínicas (Paraguay), estado civil casada y de ocupación estudiante. Consulta por lesiones en piel a nivel de la cadera en el Servicio de Dermatología. El cuadro inicia hace 4 años refiere una lesión que inicia a nivel de flanco derecho eritematoso, no pruriginosa, no dolorosa; que no aumenta de tamaño; corre el tiempo y la lesión cicatriza; la lesión tiene las siguientes características: Mancha hipocrómica de límites netos y bordes irregulares. La mayor de 3 por 15 cm de diámetro, lineal en cadera de lado derecho; los menores de 5 cm de diámetro, algunas centradas con pápulas atróficas de 0,3-0,5 cm de diámetro en la misma zona y cara interna y posterior de muslo derecho (Figura 1).
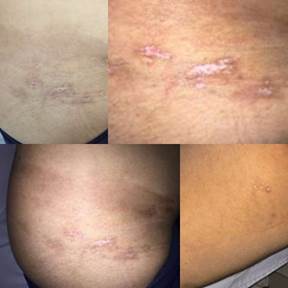
Máculas hipocrómicas de límites netos y bordes irregulares de 0,5-7,5 cm de diámetro confluentes en dorso de brazo derecho. Artralgias a nivel de codos, muñecas, tobillos; que ceden con analgésicos. Refiere fotosensibilidad. Niega alopecia, reflujo gastroesofágico. Niega Raynaud, ojo seco, boca seca, rigidez matutina, aftas, disnea y disfagia. Dentro de los antecedentes patológicos familiares presenta la madre con diagnóstico de gota y padre con artritis reumatoide, ambos con tratamiento irregular.
Con relación a los hábitos ginecológicos de la paciente, presenta un ciclo menstrual cada 28 días con una duración de 3-4 días, presentó un antecedente de aborto espontáneo a las 14 semanas de gestación. Al examen físico, la paciente presentó cifras de presión arterial de 110/80 mmHg; una frecuencia cardiaca de 72 latidos por minuto, una frecuencia respiratoria de 18 respiraciones por minuto y un peso 69,5 kg.
Al examen físico del aparato cardiovascular se halló un ritmo regular; R1 R2 normofonéticos, no soplos. Aparato respiratorio con semiología normal. Aparato digestivo se presenta abdomen blando, depresible, no doloroso, no se palpa hígado, no bazo, fosas lumbares no ocupadas, puño percusión negativo. No se observó alteración en ganglios al examen del sistema hemolinfopoyetico. El sistema nervioso central y periférico sin particularidades. Sin signos de sinovitis ni limitación de movimientos en el sistema osteoartromuscular. No se palpa tiroides en cabeza y cuello. Se observa lesión en región fosa iliaca derecha de bordes netos 10 x 10 cm, fondo hipocrómico y bordes hipercrómicos. No alopecia.
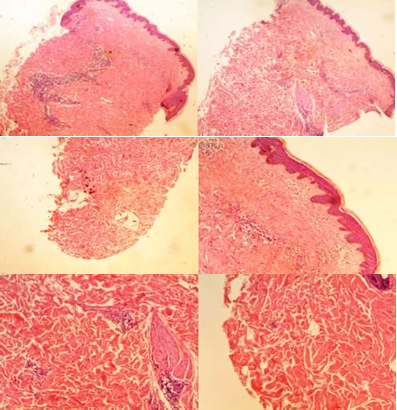
Se solicitó un laboratorio incluyendo hemograma, complemento, anticuerpos IgM e IgG para distintos agentes, anticuerpos anticentrómero y antiSCL70, y orina simple. Se realizó biopsia de piel con la siguiente descripción: Macroscopía: se remiten en el mismo envase dos cilindros cutáneos de 2 mm de eje mayor cada uno. Se incluyen totalmente y en conjunto para estudio histopatológico. Microscopía: en ambas tomas epidermis de espesor preservado, con hiperpigmentación basal.
Ausencia de rasgos liquenoides. Hiperqueratosis ortoqueratósicas y telangiectasias en dermis superficial con moderado infiltrado inflamatorio crónico perivascular de mononucleares. Aumento de espesor de la dermis por incremento del depósito de colágeno con esclerosis. No se observan unidades pilosebáceas en esta toma y los anejos sudoríparos carecen de dermis adventicial encontrándose levemente atróficos. Hiperplasia de piloerectores. Ausencia de hallazgos vasculares Con lo que se llega al diagnóstico definitivo de Morfea (Figura 2).
Se realiza examen ocular con resultado normal. Se pidió para iniciar Hidroxicloroquina. Dentro de los diagnósticos diferenciales se estableció a la esclerosis sistémica ya que es importante primero establecer un diagnóstico diferencial con esclerodermia sistémica ante un paciente con signos de esclerosis o atrofia debido al pronóstico de esta. La ausencia de fenómeno de Raynaud y/o síntomas sistémicos, afectación de la piel en zonas acrales y la preservación de la microcirculación periungueal indican más una esclerodermia localizada9. El síndrome de superposición caracterizada por la expresión de dos o más enfermedades definidas siendo las asociaciones más frecuentes entre lupus, esclerosis, polimiositis y artritis. En este caso en particular, la presencia de fotosensibilidad y artralgias podrían sugerir sospecha de superposición, sin embrago, los estudios laboratoriales alejan esta probabilidad10. La enfermedad mixta del tejido conectivo: cuando se observan signos que corresponden a diversas enfermedades sin que se pueda definir una entidad concreta, o pueden tener un patrón más definido asociado a la presencia de anti-RNP llamándose entonces enfermedad mixta del tejido conectivo, que puede evolucionar y diferenciarse en otras enfermedades autoinmunes, principalmente lupus, esclerodermia o miositis y la escleredema o enfermedad de Buschke donde las lesiones cutáneas manifiestas en esta enfermedad son áreas extensas induradas que no delimitan placas, sin una limitación entre piel sana y piel afecta, no se presentan áreas hiperpigmentadas y de atrofia como es característico de la morfea11.
Se inició tratamiento con Hidroxicloroquina 200 mg/día, Metotrexato 15 mg/semanal, Ácido fólico 5 mg/semanal, Vitamina C: 2g/día, Protector solar FPS 40. Vitamina E. Crema humectante con Vitamina A. Corticoides local (crema). Tras la evolución, se suspendió Metotrexato por embarazo a las 3 semanas de gestación. Trae a la consulta sus resultados de laboratorio de rutina de sangre y orina. No refiere artralgias, disfagia, fotosensibilidad, alopecia, fiebre y rash malar. Boca seca, ojo seco: negativo. Disnea: negativo. Raynaud: negativo Al aparato cardiovascular se halló R1 y R2 normofonéticos no soplos. Aparato Respiratorio con murmullo vesicular conservado, no rales. Sistema Osteoartromuscular no presenta sinovitis. En piel se observa lesión en fosa iliaca derecha, bordes netos sobreelevados, hipertróficos, centro hipotrófico, no doloroso. Se indica tratamiento con Hidroxicloroquina 200 mg/día. Vitamina C 2g/día. Protector solar FPS 40. Vitamina E. Crema humectante con Vitamina A. Se solicitó perfil SAF y perfil ENA. Tras evolución favorable, sin datos de actividad de la enfermedad clínica y laboratorial, se decidió continuar con el tratamiento actual (Tabla 1).
Tabla 1: Laboratorio. Realizado en el Hospital de Clínicas, San Lorenzo.
| Hemoglobina | 10,5 g/dl |
| Hematocrito | 31,70% |
| VCM | 90 fl |
| HCM | 30 pg |
| Glóbulos Blancos | 7200/mm3 |
| Linfocitos | 29% |
| Neutrófilos | 60% |
| VSG | 40 mm |
| ANA | Negativo |
| Anticuerpos anti-DNA | Negativo |
| C3 | 111 mg/dl |
| C4 | 19,1 mg/dl |
| Chagas IgG | Negativo |
| Chagas IgM | Negativo |
| CMV IgG | Negativo |
| CMV IgM | Negativo |
| Rubeola IgG | Positivo |
| Rubeola IgM | Negativo |
| Herpes simple 1 IgG | Positivo |
| Herpes simple 1 IgM | Negativo |
| Herpes simple 2 IgG | Positivo |
| Herpes simple 2 IgM | Negativo Ac |
| Anti SCL 70 | Negativo |
| Anti centrómero IgG | Negativo |
| Toxoplasmosis IgG | Positivo |
| Toxoplasmosis IgM | Negativo |
| Anticuerpos anti-Antígenos Extractables (ENA) | Negativo |
| Anticuerpos anti-Cardiolipina IgG | 2 GPL U/ml |
| Anticuerpos anti-Cardiolipina IgM | 4 GPL U/ml |
Discusión
De acuerdo con la clasificación de presentación clínica de la LS, podemos decir que la más frecuente es la morfea circunscripta. La paciente presenta una LS de subtipo generalizada con placas coalescentes por presentar más de 4 placas en más de dos o tres regiones anatómicas del cuerpo; de presentación más infrecuente y que muchas veces resulta difícil de diferenciar de la esclerosis sistémica; la ausencia del fenómeno de Raynaud, la afectación del lecho ungual o la esclerodactilia, alejan la posibilidad de una esclerosis sistémica; estando ausentes los tres en este caso. En la LS tampoco se demuestran anticuerpos antirribonucleoproteína, anticentrómero, ni anti Scl-70; en la paciente estos dos últimos en la serología resultaron negativos. El diagnóstico es clínico y se confirma mediante estudio histopatológico12.
La morfea generalizada es mucho más frecuente en mujeres. Las placas son ligeramente inflamatorias, bien definidas, engrosadas, adheridas a planos profundos, fascia y músculos. De asiento mucho más frecuente en tronco y extremidades. El establecimiento de la esclerosis es gradual y relativamente rápido durante un periodo de unos cuantos meses. La morfea generalizada es diferente de la esclerosis sistémica, los pacientes pueden tener esclerodactilia pero no presentan ulceraciones, resorciones falángeas, cambios en los capilares del lecho ungueal o el fenómeno de Raynaud, lo cual sí ocurre en la esclerosis sistémica13.
La apariencia de las lesiones cambia a lo largo del tiempo y resulta útil preguntar al paciente al respecto. Las lesiones al comienzo tienen bordes eritemato-violáceo que circunscriben las áreas de esclerosis, difícilmente son diagnosticados en esta etapa; luego desarrollan una esclerosis central y eventualmente la inflamación disminuye y el tejido es reemplazado por hiperpigmentación post-inflamatoria y con grados variables de esclerosis central, y frecuentemente suele haber pérdida de anexos cutáneos (usualmente de los folículos pilosos, pero se conservan las glándulas sudoríparas). A la histopatología, que varía según la etapa de la enfermedad y el sitio donde se tome la biopsia, el patrón grave de esclerosis y la presencia de infiltrado inflamatorio severo pueden ser una indicación para una terapia agresiva (ej: inmunosupresores sistémicos), especialmente en la morfea generalizada14-16.
La paciente refirió dolores articulares y fotosensibilidad que mejoraron tras el tratamiento sistémico con los inmunosupresores. Se han reportado artralgias asociadas en 44 % de pacientes con morfea en placa y en 40 % de pacientes con morfea generalizada. Estudios recientes señalan la presencia de retrasos significativos en el diagnóstico de estos pacientes, esto podría deberse a la subestimación de las lesiones y su presentación clínica heterogénea. Es importante recalcar que las lesiones son más receptivas al tratamiento mientras estén activas14.
La esclerosis sistémica y la morfea son dos entidades clínicas distintas y que pueden compartir algunas características, como los hallazgos histopatológicos en la piel y posible presencia de autoanticuerpos antinucleares (ANA), esto induce a creer que estas podrían representar dos extremos del espectro de una única enfermedad. En un estudio realizado en Italia de una cohorte de 330 pacientes con esclerodermia sistémica, 8 (2,4 %) pacientes tenían también morfea, de los cuales 6 pacientes mujeres (1,8 %) tenían historia de esclerodermia localizada anterior al diagnóstico de esclerosis sistémica17. Aún no existen pruebas diagnósticas laboratoriales para la morfea, pero se encuentra un ANA positivo en el 42 a 73 % en los sujetos con morfea, más frecuentemente hallado en etapas tempranas de la enfermedad, y esta ha sido asociada con un riesgo aumentado de complicaciones extracutáneas18, pero en la paciente tuvo ANA negativos al momento del diagnóstico y en los controles siguientes.
Una práctica bien aceptada tanto por reumatólogos y dermatólogos es la aplicación de un tratamiento sistémico para el manejo de las formas severas e incapacitantes de la morfea, que incluye también las lesiones que sean resistentes al tratamiento tópico o que estén en una localización inaceptable (como el rostro y cuero cabelludo o transversal a una articulación), compromiso subcutáneo profundo o lesiones extensas mayores a una sola lesión en placa19. En este caso, se utilizó al principio metotrexate y la hidroxicloroquina, para la utilización de este último medicamento se realizó un examen ocular previo. El metotrexate en combinación con los corticosteroides y la terapia con rayos UV tienen la evidencia científica suficiente para sustentar el tratamiento20. El metotrexate tuvo que ser suspendido debido a que este se encuentra contraindicada su utilización durante el embarazo debido al potencial abortivo y malformativo21, quedando solo la hidroxicloroquina con el resto de las medicaciones. El tratamiento sistémico en la paciente fue complementado con corticoesteroideos tópicos, protector solar, ácido fólico y vitaminas A y E.

